A SMILES (Simplified molecular-input line-entry system) string is a string that represents a chemical structure using ASCII characters. For example, water (\$H_2O\$) can be written in SMILES as H-O-H.
However, for simplicity, the single bonds (-) and hydrogen atoms (H) are frequently omitted. Thus, a molecules with only single bonds like n-pentane (\$CH_3CH_2CH_2CH_2CH_3\$) can be represented as simply CCCCC, and ethanol (\$CH_3CH_2OH\$) as CCO or OCC (which atom you start from does not matter).
n-pentane:n-pentane
{kind=link}
ethanol:ethanol
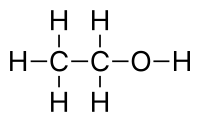{kind=link}
In SMILES, double bonds are represented with = and triple bonds with #. So ethene:
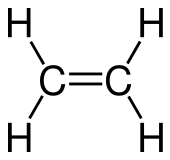{kind=link}
can be represented as C=C, and hydrogen cyanide:
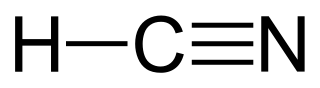{kind=link}
can be represented as C#N or N#C.
SMILES uses parentheses when representing branching:
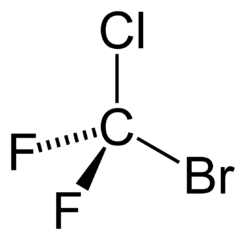{kind=link}
Bromochlorodifluoromethane can be represented as FC(Br)(Cl)F, BrC(F)(F)Cl, C(F)(Cl)(F)Br, etc.
For rings, atoms that close rings are numbered:
{kind=link}
First strip the H and start from any C. Going round the ring, we get CCCCCC. Since the first and last C are bonded, we write C1CCCCC1.
Use this tool: https://pubchem.ncbi.nlm.nih.gov/edit3/index.html to try drawing your own structures and convert them to SMILES, or vice versa.
Task
Your program shall receive two SMILES string. The first one is a molecule, the second is a substructure (portion of a molecule). The program should return true if the substructure is found in the molecule and false if not. For simplicity, only above explanation of SMILES will be used (no need to consider stereochemistry like cis-trans, or aromaticity) and the only atoms will be:
OCNF
Also, the substructure do not contain H.
Examples
CCCC C
true
CCCC CC
true
CCCC F
false
C1CCCCC1 CC
true
C1CCCCC1 C=C
false
COC(C1)CCCC1C#N C(C)(C)C // substructure is a C connected to 3 other Cs
true
COC(C1)CCCCC1#N COC1CC(CCC1)C#N // SMILES strings representing the same molecule
true
OC(CC1)CCC1CC(N)C(O)=O CCCCO
true
OC(CC1)CCC1CC(N)C(O)=O NCCO
true
OC(CC1)CCC1CC(N)C(O)=O COC
false
Shortest code wins. Refrain from using external libraries.
1 Answer 1
Mathematica, 31 bytes
MoleculeContainsQ@@Molecule/@#&
Takes input as a list of 2 strings (the source and the pattern molecules).
As you could guess, this checks if the first molecule (parsed via Molecule) contains the second one by using the MoleculeContainsQ function.
This doesn't seem to work in the online interpreter on TIO; I'm not sure what I am doing wrong. It works on my local machine, though. Of course, this is not using an external library: it's completely built-in functionality!
-
1\$\begingroup\$ i'm looking for answers that do some parsing, but as your answer uses built-in functionality, i guess you've found a loophole! +1 \$\endgroup\$kuantumleap123– kuantumleap1232020年06月12日 06:25:03 +00:00Commented Jun 12, 2020 at 6:25
containsbuiltin. \$\endgroup\$COC(C1)CCCC1C#Ncan't be pasted to the tool you've linked (it automatically changes toCOC1CC(CCC1)C#N..) Also, wouldCOC(C1)CCCCC1#NwithCCCCCCresult in truthy, since it does contain a substructure of six subsequenceC-atoms (the entire circle(C1)CCCCC1, and the additional branch to aCinCOC)? \$\endgroup\$COC(C1)CCCCC1#NwithCCCCCCwill be truthy.COC(C1)CCCCC1#NwithCOC1CC(CCC1)C#Nwill be truthy. \$\endgroup\$