Machine learning with physicochemical relationships: solubility prediction in organic solvents and water
- PMID: 33188226
- PMCID: PMC7666209
- DOI: 10.1038/s41467-020-19594-z
Machine learning with physicochemical relationships: solubility prediction in organic solvents and water
Abstract
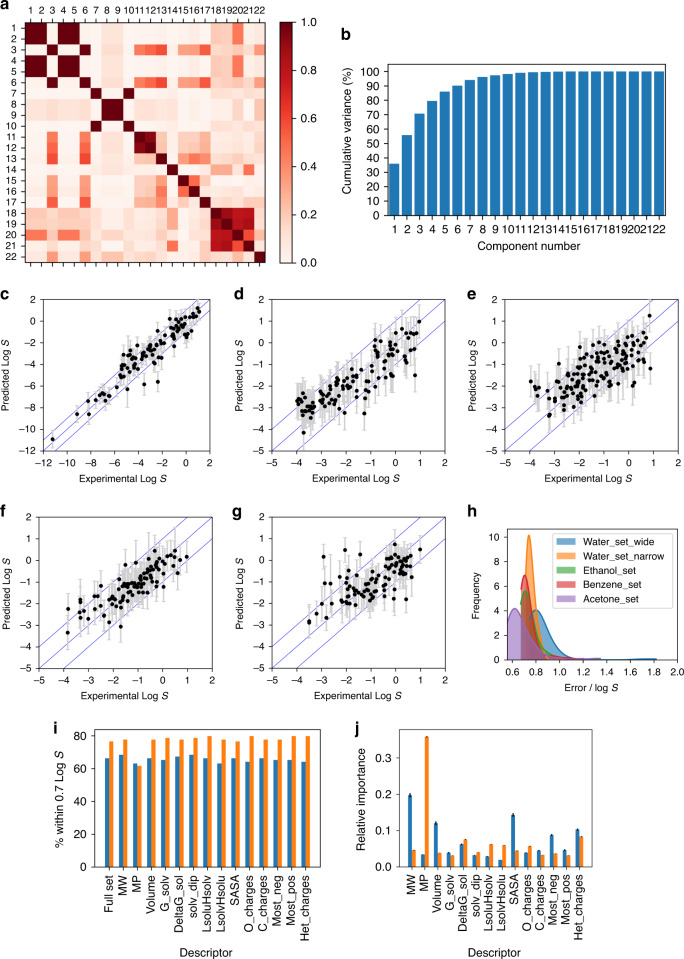
Solubility prediction remains a critical challenge in drug development, synthetic route and chemical process design, extraction and crystallisation. Here we report a successful approach to solubility prediction in organic solvents and water using a combination of machine learning (ANN, SVM, RF, ExtraTrees, Bagging and GP) and computational chemistry. Rational interpretation of dissolution process into a numerical problem led to a small set of selected descriptors and subsequent predictions which are independent of the applied machine learning method. These models gave significantly more accurate predictions compared to benchmarked open-access and commercial tools, achieving accuracy close to the expected level of noise in training data (LogS ± 0.7). Finally, they reproduced physicochemical relationship between solubility and molecular properties in different solvents, which led to rational approaches to improve the accuracy of each models.
Conflict of interest statement
The authors declare no competing interests.
Figures
{kind=link}
{kind=link}
{kind=link}
References
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources