Meiotic sex in Chagas disease parasite Trypanosoma cruzi
- PMID: 31481692
- PMCID: PMC6722143
- DOI: 10.1038/s41467-019-11771-z
Meiotic sex in Chagas disease parasite Trypanosoma cruzi
Abstract
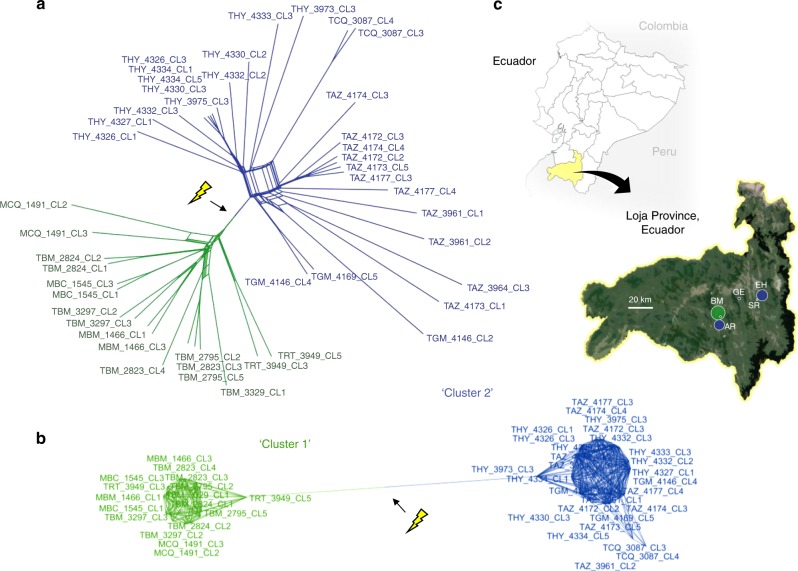
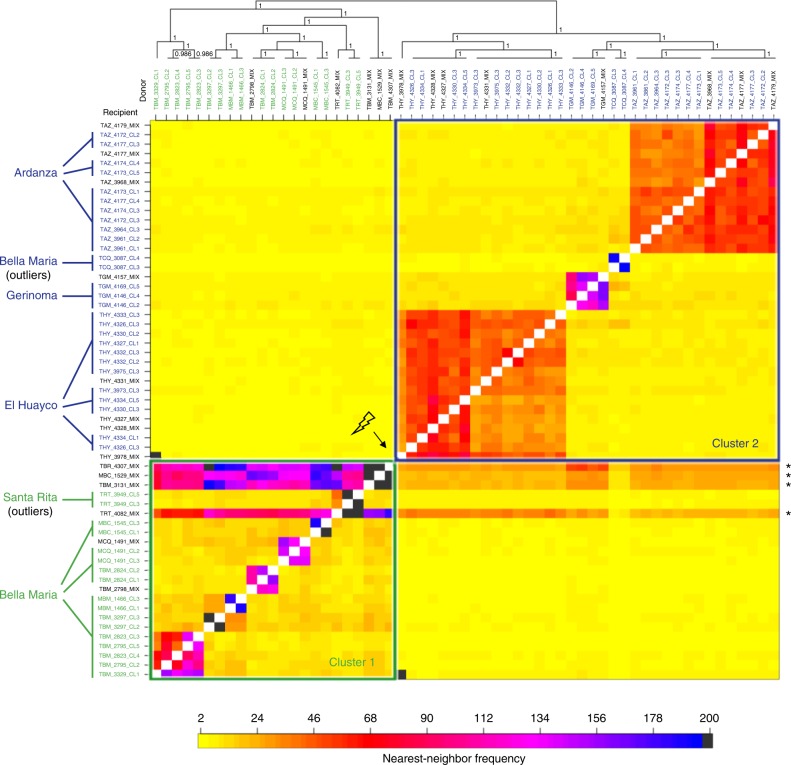
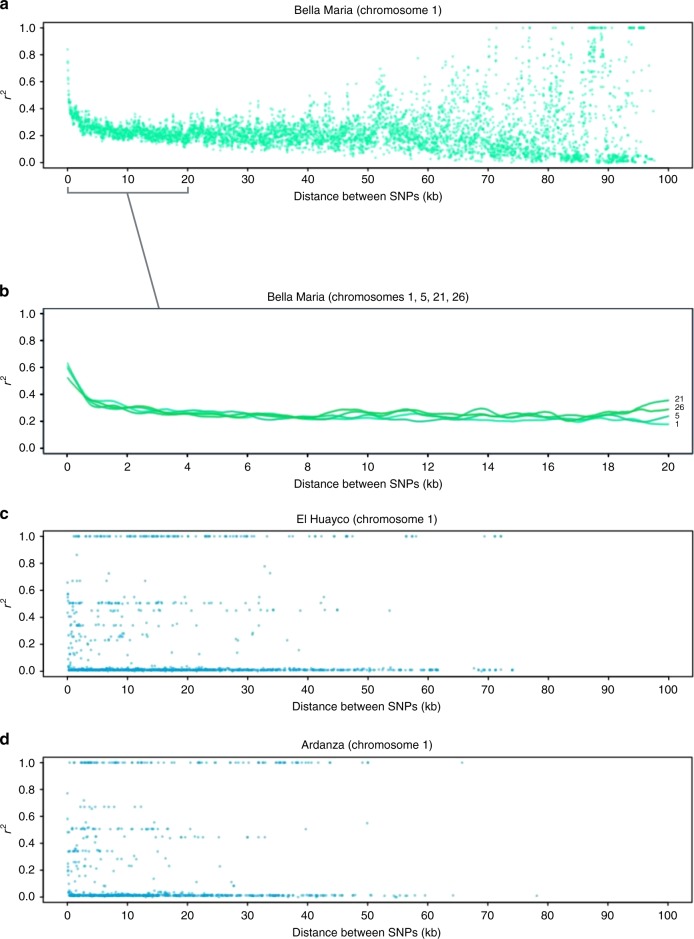
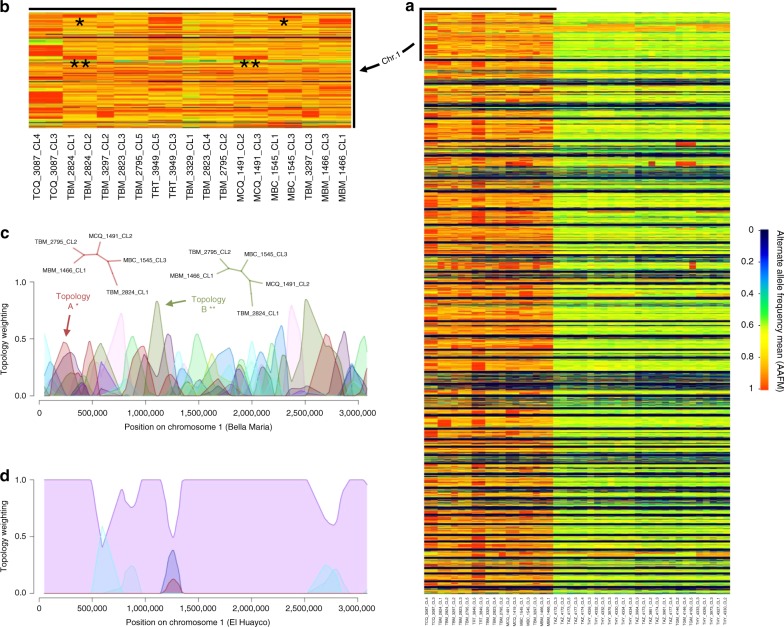
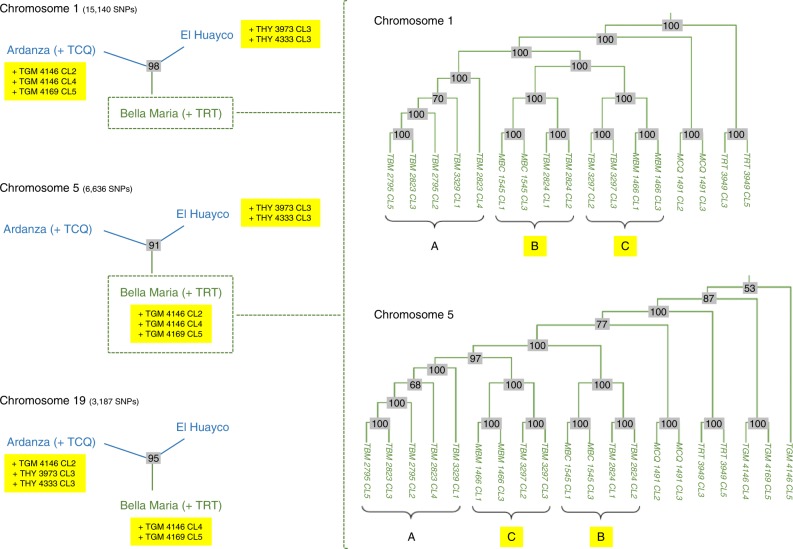
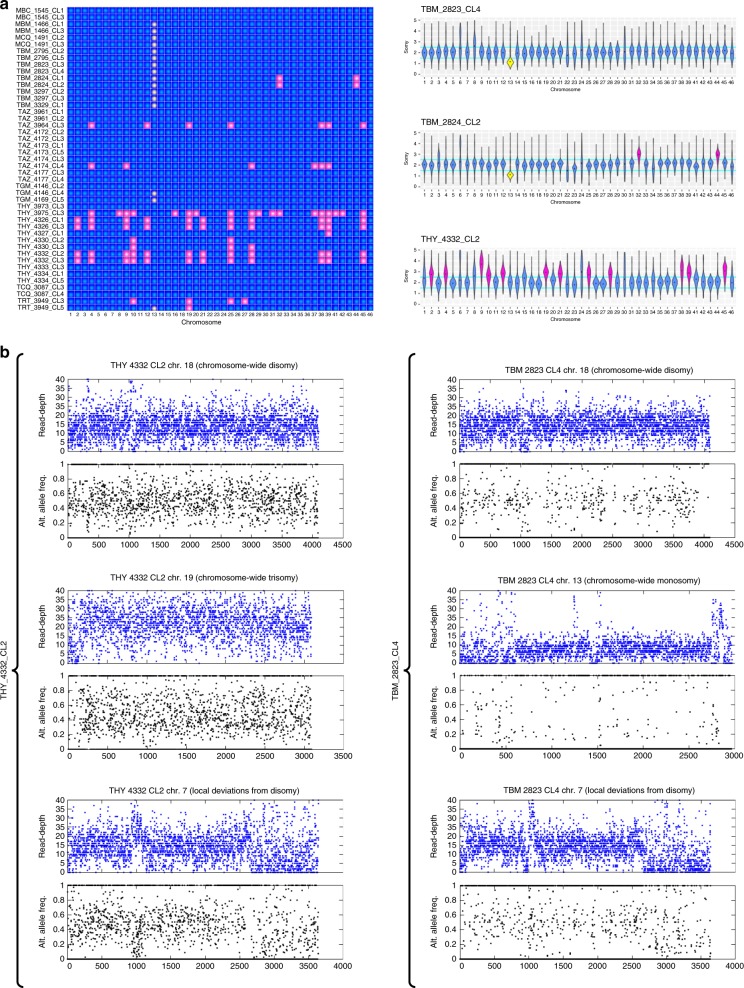
Genetic exchange enables parasites to rapidly transform disease phenotypes and exploit new host populations. Trypanosoma cruzi, the parasitic agent of Chagas disease and a public health concern throughout Latin America, has for decades been presumed to exchange genetic material rarely and without classic meiotic sex. We present compelling evidence from 45 genomes sequenced from southern Ecuador that T. cruzi in fact maintains truly sexual, panmictic groups that can occur alongside others that remain highly clonal after past hybridization events. These groups with divergent reproductive strategies appear genetically isolated despite possible co-occurrence in vectors and hosts. We propose biological explanations for the fine-scale disconnectivity we observe and discuss the epidemiological consequences of flexible reproductive modes. Our study reinvigorates the hunt for the site of genetic exchange in the T. cruzi life cycle, provides tools to define the genetic determinants of parasite virulence, and reforms longstanding theory on clonality in trypanosomatid parasites.
Conflict of interest statement
The authors declare no competing interests.
Figures
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
References
-
- Coura, J. R. & Viñas, P. A. Chagas disease: a new worldwide challenge. Nature.465, S6–S7 (2010). - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous