Evaluation of ultra-low input RNA sequencing for the study of human T cell transcriptome
- PMID: 31186477
- PMCID: PMC6559993
- DOI: 10.1038/s41598-019-44902-z
Evaluation of ultra-low input RNA sequencing for the study of human T cell transcriptome
Abstract
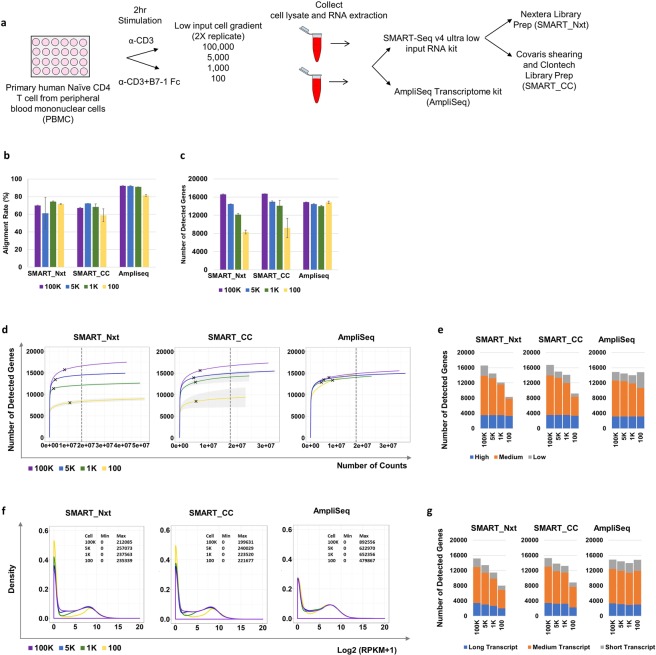
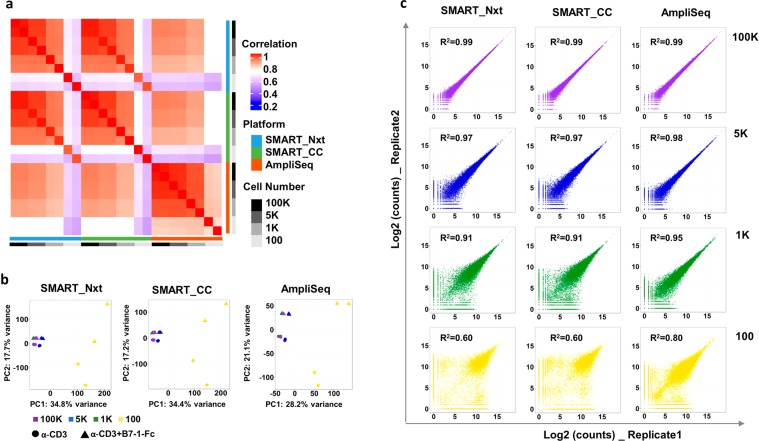
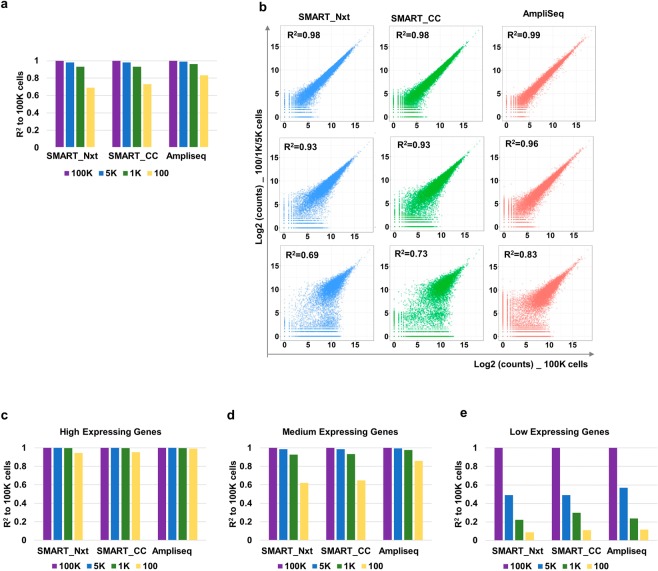
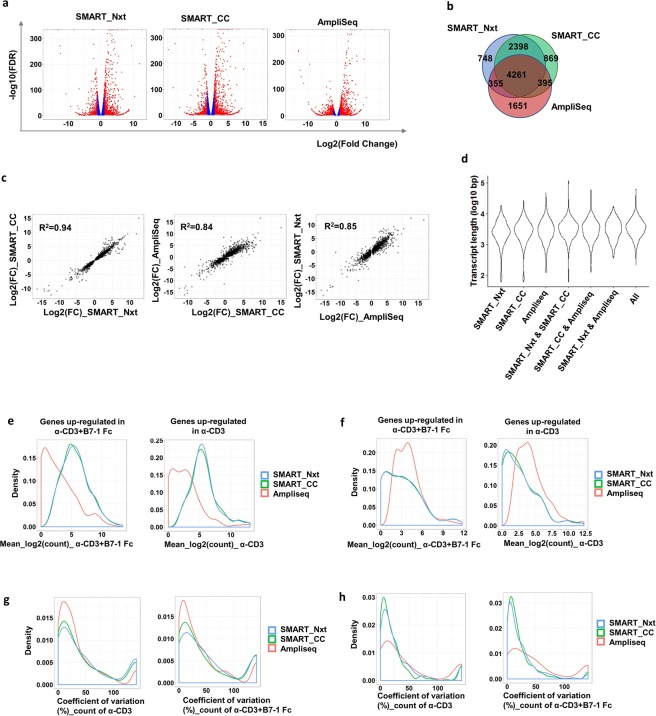
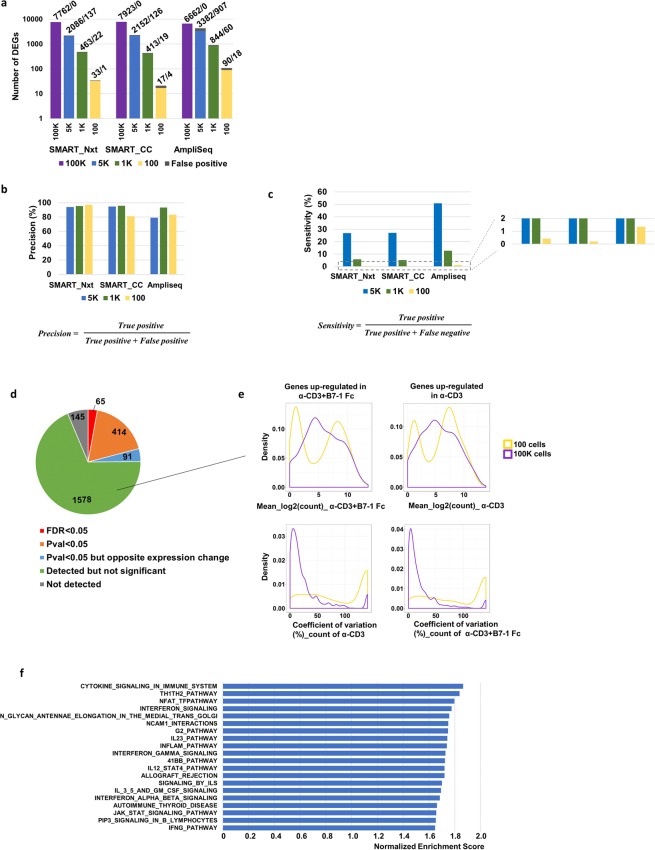
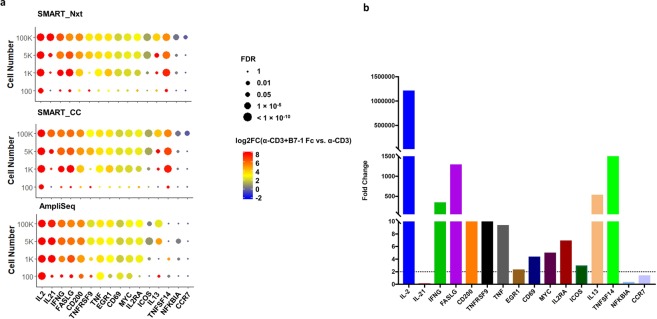
Deeper understanding of T cell biology is crucial for the development of new therapeutics. Human naïve T cells have low RNA content and their numbers can be limiting; therefore we set out to determine the parameters for robust ultra-low input RNA sequencing. We performed transcriptome profiling at different cell inputs and compared three protocols: Switching Mechanism at 5' End of RNA Template technology (SMART) with two different library preparation methods (Nextera and Clontech), and AmpliSeq technology. As the cell input decreased the number of detected coding genes decreased with SMART, while stayed constant with AmpliSeq. However, SMART enables detection of non-coding genes, which is not feasible for AmpliSeq. The detection is dependent on gene abundance, but not transcript length. The consistency between technical replicates and cell inputs was comparable across methods above 1 K but highly variable at 100 cell input. Sensitivity of detection for differentially expressed genes decreased dramatically with decreased cell inputs in all protocols, support that additional approaches, such as pathway enrichment, are important for data interpretation at ultra-low input. Finally, T cell activation signature was detected at 1 K cell input and above in all protocols, with AmpliSeq showing better detection at 100 cells.
Conflict of interest statement
Wang J., Rieder S., Hayes S., Halpin R., de los Reyes M., Shrestha Y., Kolbeck R., Raja R. are full time employees and shareholders of AstraZeneca plc. Wu J has been a full time employee and shareholder of AstraZeneca plc until recently.
Figures
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials