Impact of Childhood Malnutrition on Host Defense and Infection
- PMID: 28768707
- PMCID: PMC5608884
- DOI: 10.1128/CMR.00119-16
Impact of Childhood Malnutrition on Host Defense and Infection
Abstract
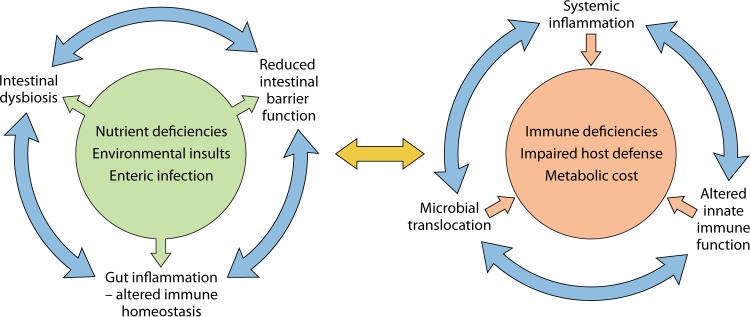
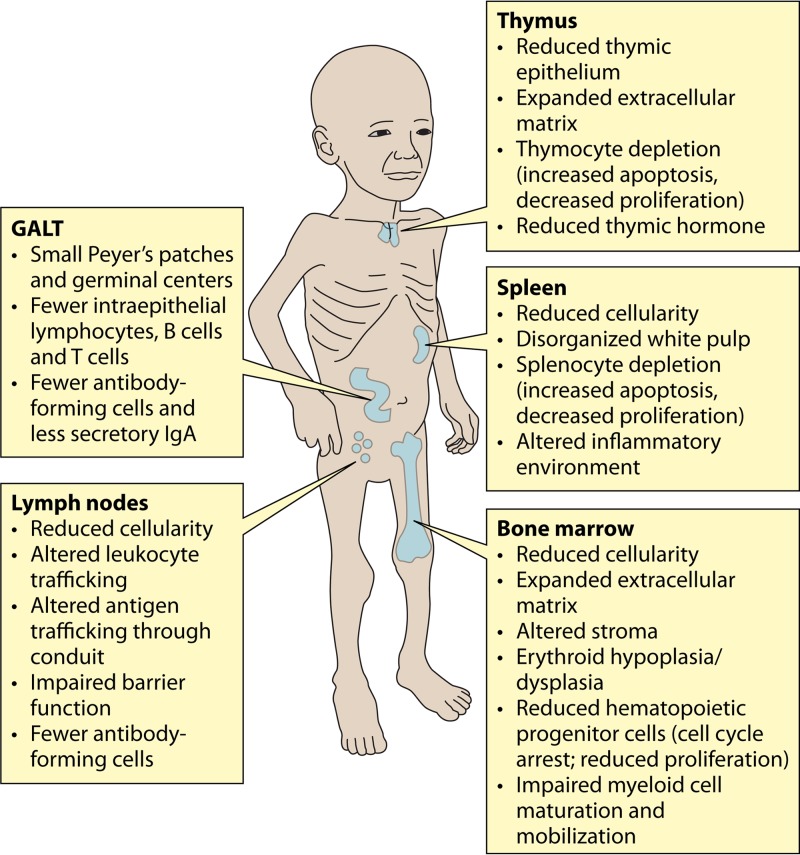
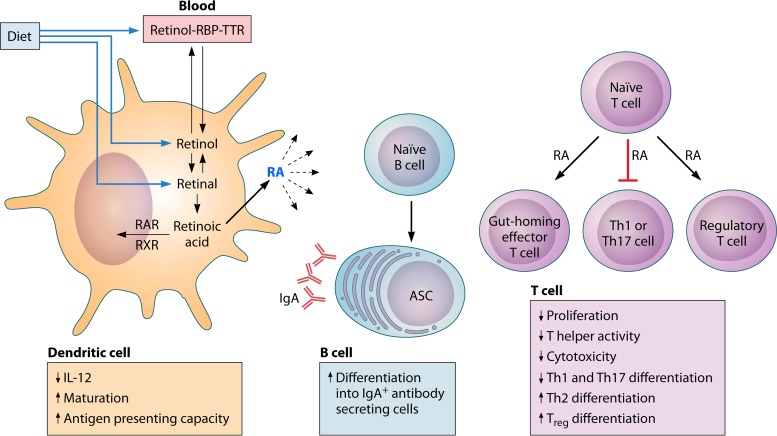
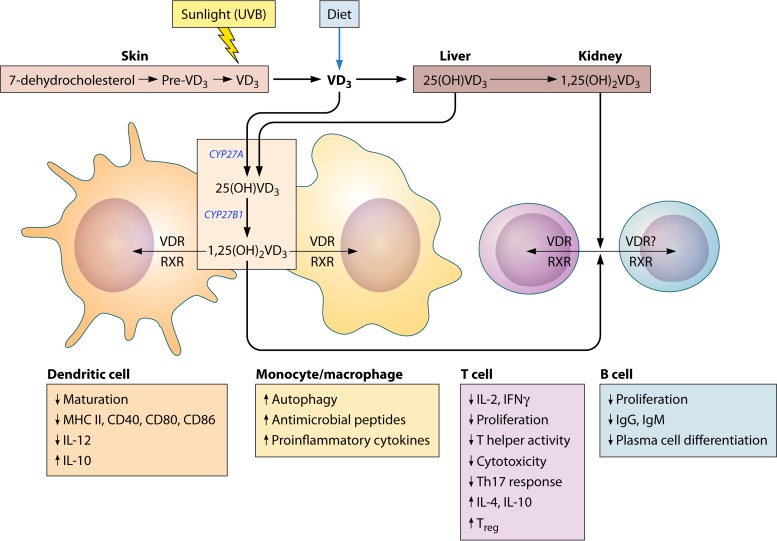
The global impact of childhood malnutrition is staggering. The synergism between malnutrition and infection contributes substantially to childhood morbidity and mortality. Anthropometric indicators of malnutrition are associated with the increased risk and severity of infections caused by many pathogens, including viruses, bacteria, protozoa, and helminths. Since childhood malnutrition commonly involves the inadequate intake of protein and calories, with superimposed micronutrient deficiencies, the causal factors involved in impaired host defense are usually not defined. This review focuses on literature related to impaired host defense and the risk of infection in primary childhood malnutrition. Particular attention is given to longitudinal and prospective cohort human studies and studies of experimental animal models that address causal, mechanistic relationships between malnutrition and host defense. Protein and micronutrient deficiencies impact the hematopoietic and lymphoid organs and compromise both innate and adaptive immune functions. Malnutrition-related changes in intestinal microbiota contribute to growth faltering and dysregulated inflammation and immune function. Although substantial progress has been made in understanding the malnutrition-infection synergism, critical gaps in our understanding remain. We highlight the need for mechanistic studies that can lead to targeted interventions to improve host defense and reduce the morbidity and mortality of infectious diseases in this vulnerable population.
Keywords: Mycobacterium tuberculosis; host defense; immunology; infectious disease; malaria; malnutrition; micronutrients; pneumonia; sepsis.
Copyright © 2017 American Society for Microbiology.
Figures
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical