Improved maize reference genome with single-molecule technologies
- PMID: 28605751
- PMCID: PMC7052699
- DOI: 10.1038/nature22971
Improved maize reference genome with single-molecule technologies
Abstract
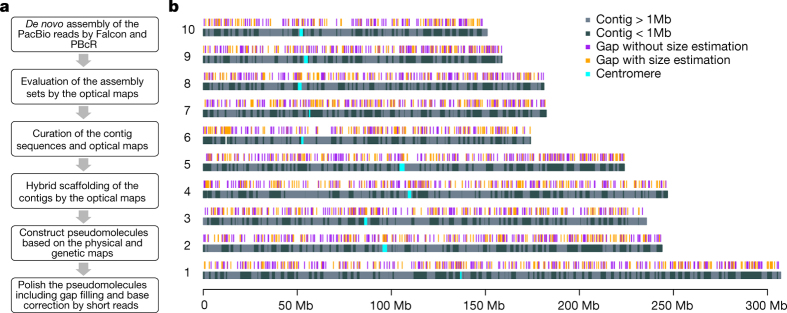
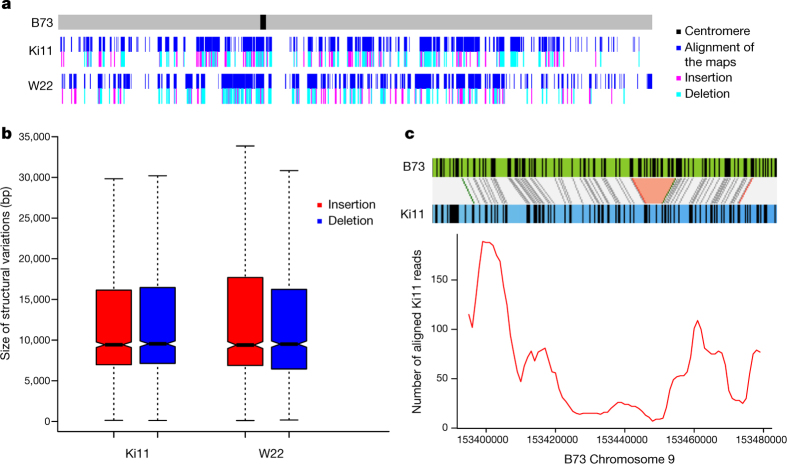
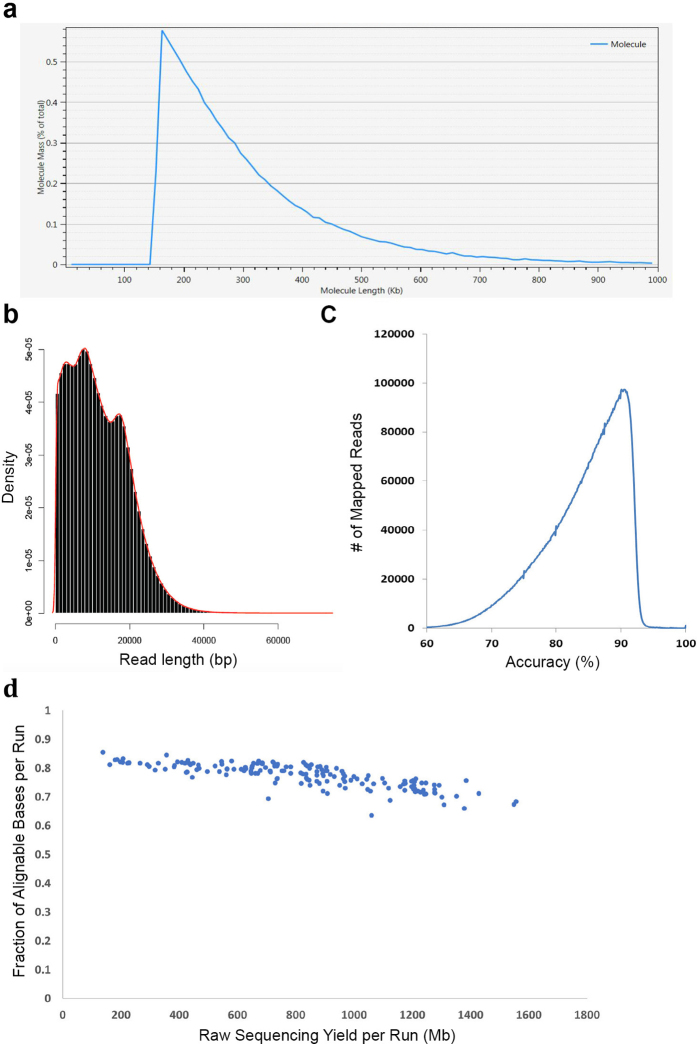
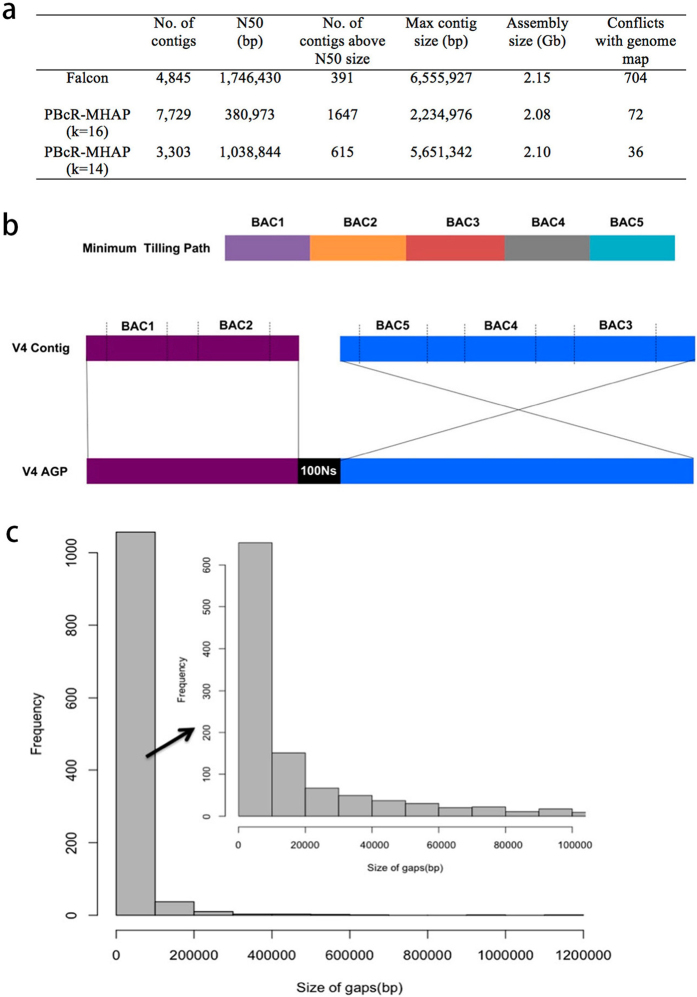
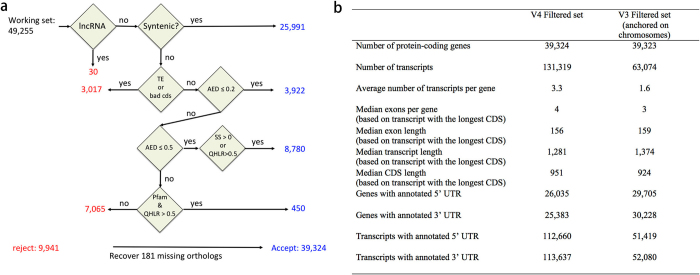
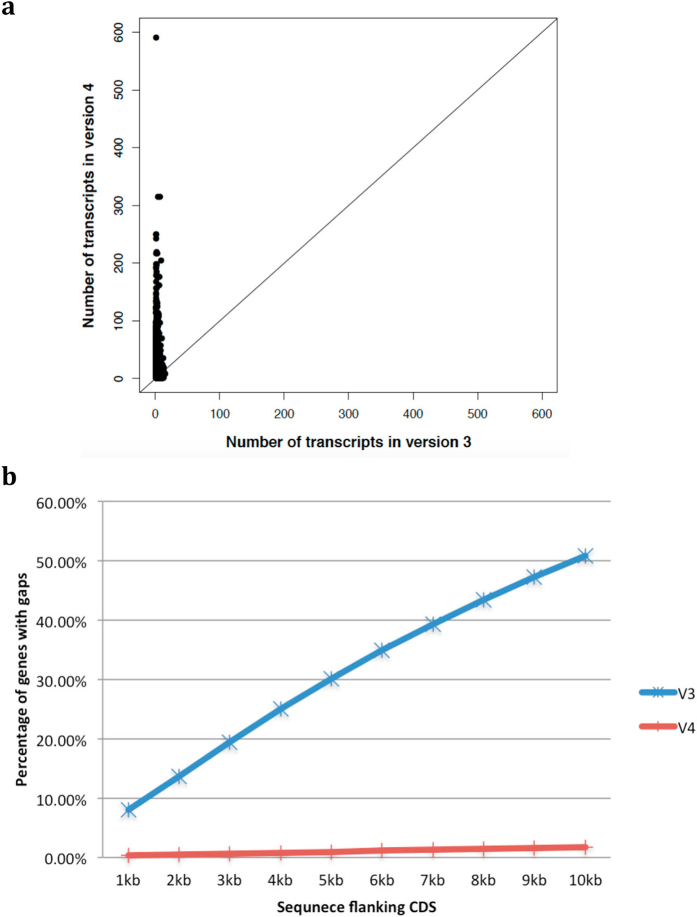
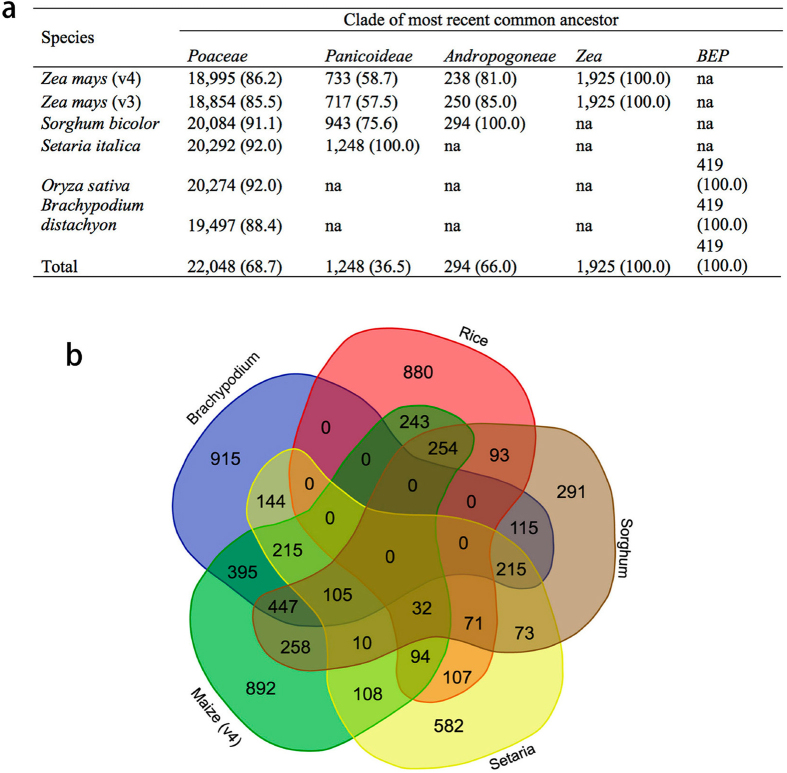
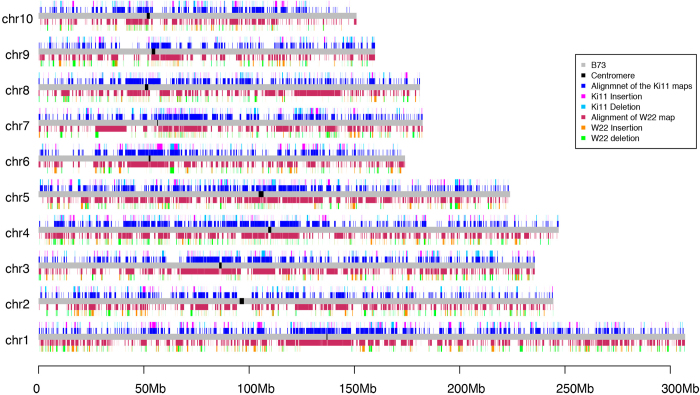
Complete and accurate reference genomes and annotations provide fundamental tools for characterization of genetic and functional variation. These resources facilitate the determination of biological processes and support translation of research findings into improved and sustainable agricultural technologies. Many reference genomes for crop plants have been generated over the past decade, but these genomes are often fragmented and missing complex repeat regions. Here we report the assembly and annotation of a reference genome of maize, a genetic and agricultural model species, using single-molecule real-time sequencing and high-resolution optical mapping. Relative to the previous reference genome, our assembly features a 52-fold increase in contig length and notable improvements in the assembly of intergenic spaces and centromeres. Characterization of the repetitive portion of the genome revealed more than 130,000 intact transposable elements, allowing us to identify transposable element lineage expansions that are unique to maize. Gene annotations were updated using 111,000 full-length transcripts obtained by single-molecule real-time sequencing. In addition, comparative optical mapping of two other inbred maize lines revealed a prevalence of deletions in regions of low gene density and maize lineage-specific genes.
Conflict of interest statement
P.P., C.-S.C. and D.R.R. are full-time employees of Pacific Biosciences. J.S., T.L. and A.H. are employees of BioNano Genomics, Inc., and own company stock options. W.R.M. has participated in Illumina sponsored meetings over the past four years and received travel reimbursement and an honorarium for presenting at these events. Illumina had no role in decisions relating to the study/work to be published, data collection and analysis of data, or the decision to publish. W.R.M. has participated in Pacific Biosciences sponsored meetings over the past three years and received travel reimbursement for presenting at these events. W.R.M. is a founder and shared holder of Orion Genomics, which focuses on plant genomics and cancer genetics. W.R.M. is an SAB member for RainDance Technologies, Inc. All other authors declare no competing financial interests.
Figures
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
References
-
- Edwards D, Batley J, Snowdon RJ. Accessing complex crop genomes with next-generation sequencing. Theor. Appl. Genet. 2013;126:1–11. - PubMed
-
- Morrell PL, Buckler ES, Ross-Ibarra J. Crop genomics: advances and applications. Nat. Rev. Genet. 2011;13:85–96. - PubMed
-
- Schnable PS, et al. The B73 maize genome: complexity, diversity, and dynamics. Science. 2009;326:1112–1115. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous