GenomeScope: fast reference-free genome profiling from short reads
- PMID: 28369201
- PMCID: PMC5870704
- DOI: 10.1093/bioinformatics/btx153
GenomeScope: fast reference-free genome profiling from short reads
Abstract
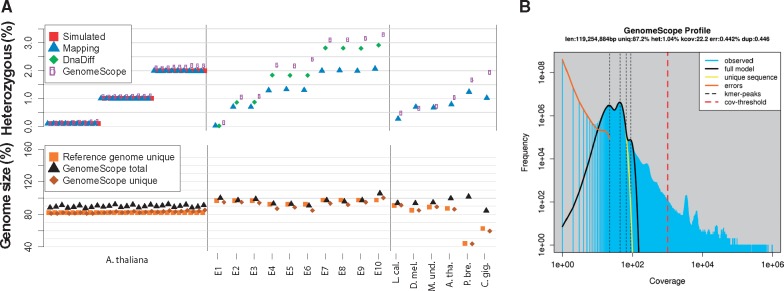
Summary: GenomeScope is an open-source web tool to rapidly estimate the overall characteristics of a genome, including genome size, heterozygosity rate and repeat content from unprocessed short reads. These features are essential for studying genome evolution, and help to choose parameters for downstream analysis. We demonstrate its accuracy on 324 simulated and 16 real datasets with a wide range in genome sizes, heterozygosity levels and error rates.
Availability and implementation: http://genomescope.org , https://github.com/schatzlab/genomescope.git .
Contact: mschatz@jhu.edu.
Supplementary information: Supplementary data are available at Bioinformatics online.
© The Author (2017). Published by Oxford University Press. All rights reserved. For Permissions, please email: journals.permissions@oup.com
Figures
{kind=link}
References
-
- Bates D.M., Watts D.G. (1988) Nonlinear Regression Analysis and Its Applications. John Wiley & Sons, Inc., New York, NY.
-
- Chikhi R., Medvedev P. (2014) Informed and automated k-mer size selection for genome assembly. Bioinformatics, 30, 31–37. - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources