SeqKit: A Cross-Platform and Ultrafast Toolkit for FASTA/Q File Manipulation
- PMID: 27706213
- PMCID: PMC5051824
- DOI: 10.1371/journal.pone.0163962
SeqKit: A Cross-Platform and Ultrafast Toolkit for FASTA/Q File Manipulation
Abstract
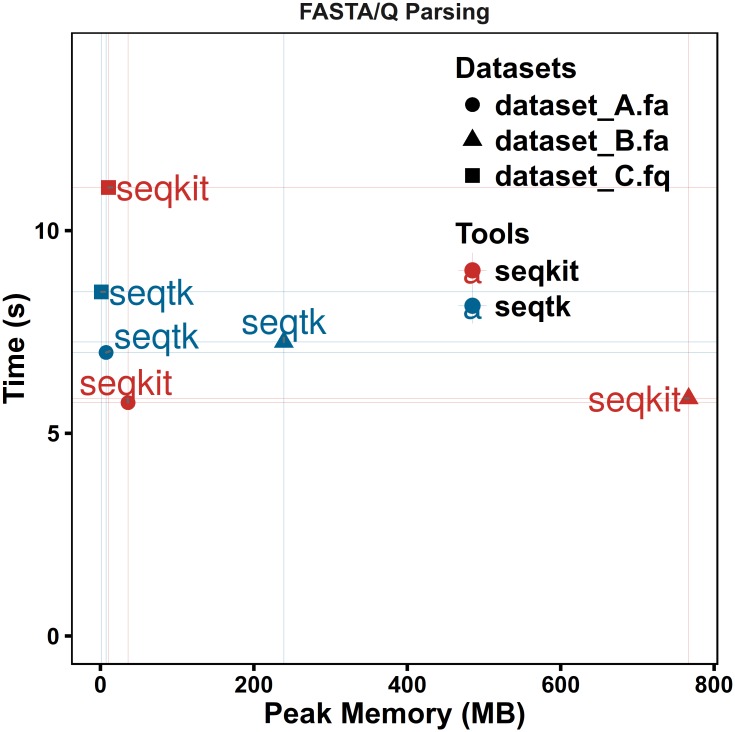
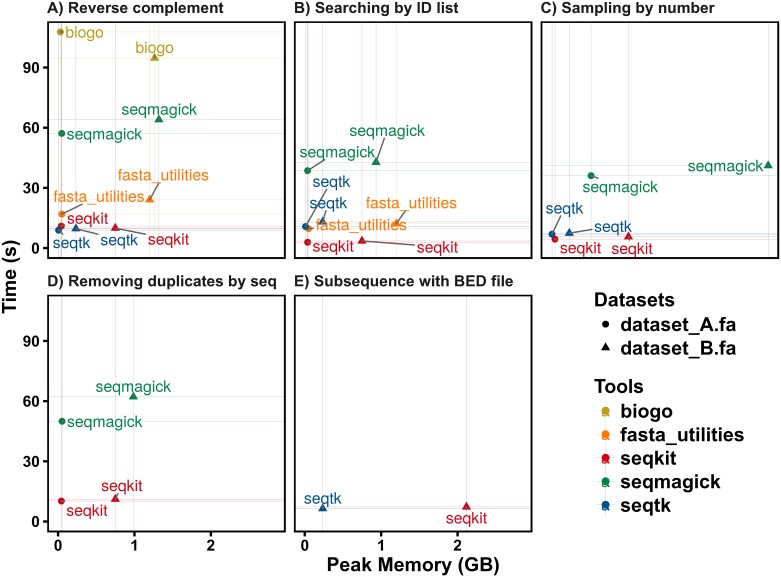
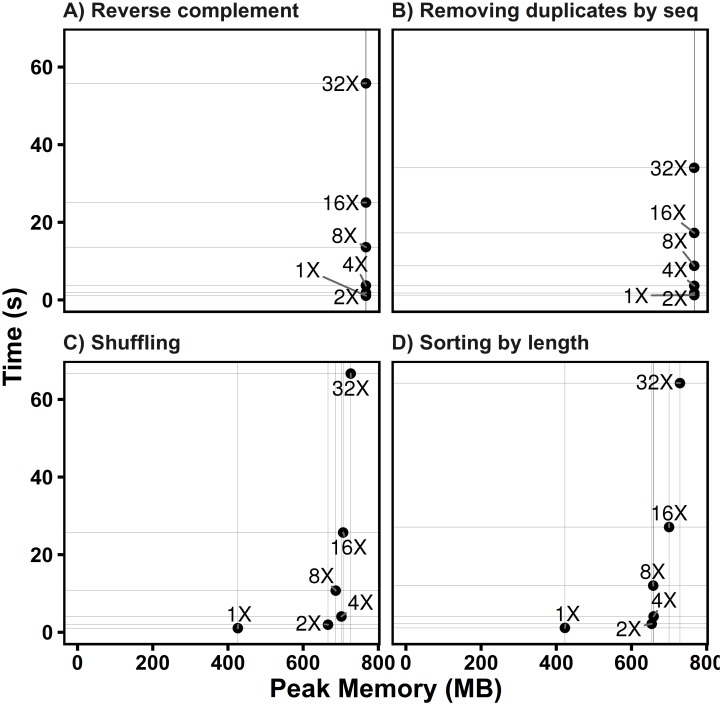
FASTA and FASTQ are basic and ubiquitous formats for storing nucleotide and protein sequences. Common manipulations of FASTA/Q file include converting, searching, filtering, deduplication, splitting, shuffling, and sampling. Existing tools only implement some of these manipulations, and not particularly efficiently, and some are only available for certain operating systems. Furthermore, the complicated installation process of required packages and running environments can render these programs less user friendly. This paper describes a cross-platform ultrafast comprehensive toolkit for FASTA/Q processing. SeqKit provides executable binary files for all major operating systems, including Windows, Linux, and Mac OSX, and can be directly used without any dependencies or pre-configurations. SeqKit demonstrates competitive performance in execution time and memory usage compared to similar tools. The efficiency and usability of SeqKit enable researchers to rapidly accomplish common FASTA/Q file manipulations. SeqKit is open source and available on Github at https://github.com/shenwei356/seqkit.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
{kind=link}
{kind=link}
{kind=link}
References
-
- Hester J. A collection of scripts developed to interact with fasta, fastq and sam/bam files. Available from: https://github.com/jimhester/fasta_utilities.
-
- FASTX-Toolkit, FASTQ/A short-reads pre-processing tools. Available from: http://hannonlab.cshl.edu/fastx_toolkit/.
-
- Shirley MD, Ma Z, Pedersen BS, Wheelan SJ. Efficient "pythonic" access to FASTA files using pyfaidx. PeerJ Preprints. 2015;3:e1196.
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources