Evolutionary and Phylogenetic Analysis of the Hepaciviruses and Pegiviruses
- PMID: 26494702
- PMCID: PMC5635594
- DOI: 10.1093/gbe/evv202
Evolutionary and Phylogenetic Analysis of the Hepaciviruses and Pegiviruses
Abstract
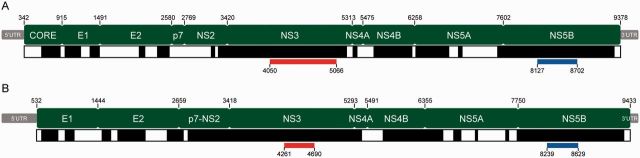
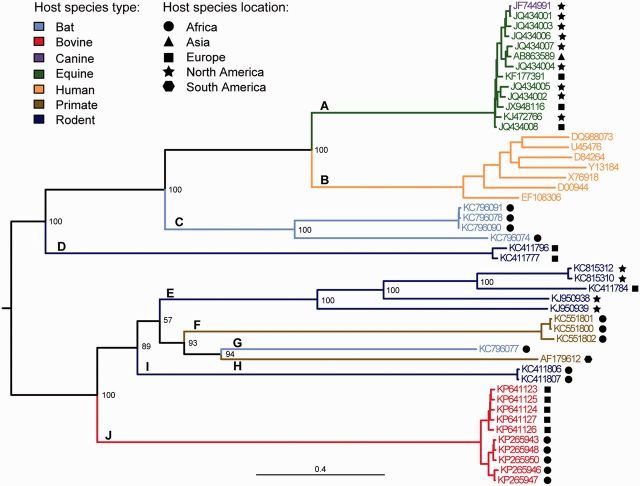
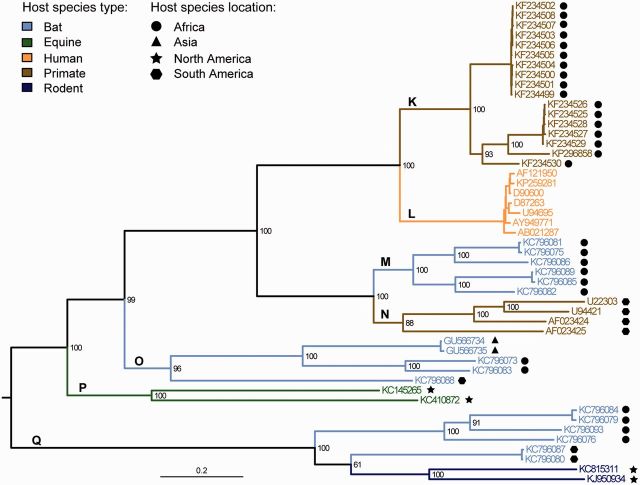
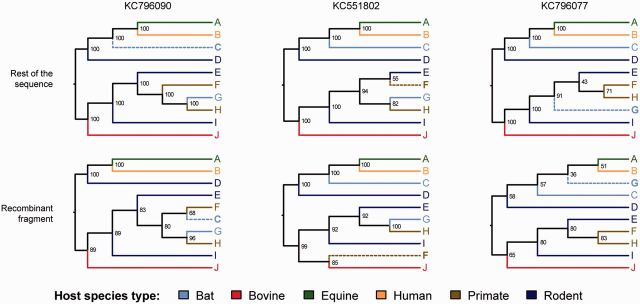
The known genetic diversity of the hepaciviruses and pegiviruses has increased greatly in recent years through the discovery of viruses related to hepatitis C virus and human pegivirus in bats, bovines, equines, primates, and rodents. Analysis of these new species is important for research into animal models of hepatitis C virus infection and into the zoonotic origins of human viruses. Here, we provide the first systematic phylogenetic and evolutionary analysis of these two genera at the whole-genome level. Phylogenies confirmed that hepatitis C virus is most closely related to viruses from horses whereas human pegiviruses clustered with viruses from African primates. Within each genus, several well-supported lineages were identified and viral diversity was structured by both host species and location of sampling. Recombination analyses provided evidence of interspecific recombination in hepaciviruses, but none in the pegiviruses. Putative mosaic genome structures were identified in NS5B gene region and were supported by multiple tests. The identification of interspecific recombination in the hepaciviruses represents an important evolutionary event that could be clarified by future sampling of novel viruses. We also identified parallel amino acid changes shared by distantly related lineages that infect similar types of host. Notable parallel changes were clustered in the NS3 and NS4B genes and provide a useful starting point for experimental studies of the evolution of Hepacivirus host-virus interactions.
Keywords: cross-species transmission; hepatitis C virus; host range; human pegivirus; parallel molecular evolution; recombination.
© The Author(s) 2015. Published by Oxford University Press on behalf of the Society for Molecular Biology and Evolution.
Figures
{kind=link}
{kind=link}
{kind=link}
{kind=link}
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources