HISAT: a fast spliced aligner with low memory requirements
- PMID: 25751142
- PMCID: PMC4655817
- DOI: 10.1038/nmeth.3317
HISAT: a fast spliced aligner with low memory requirements
Abstract
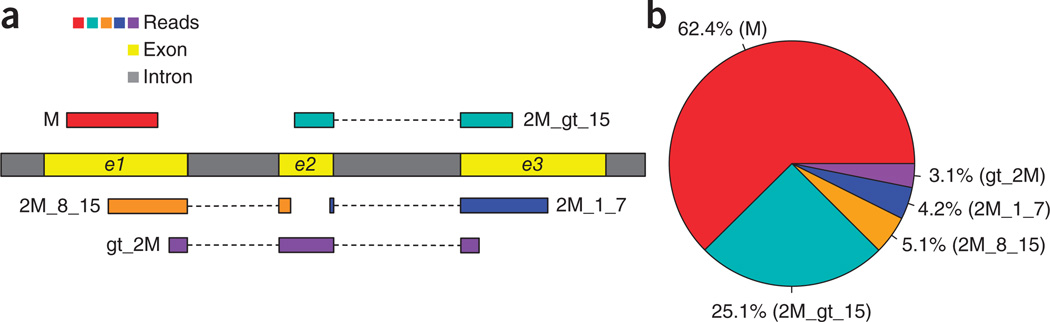
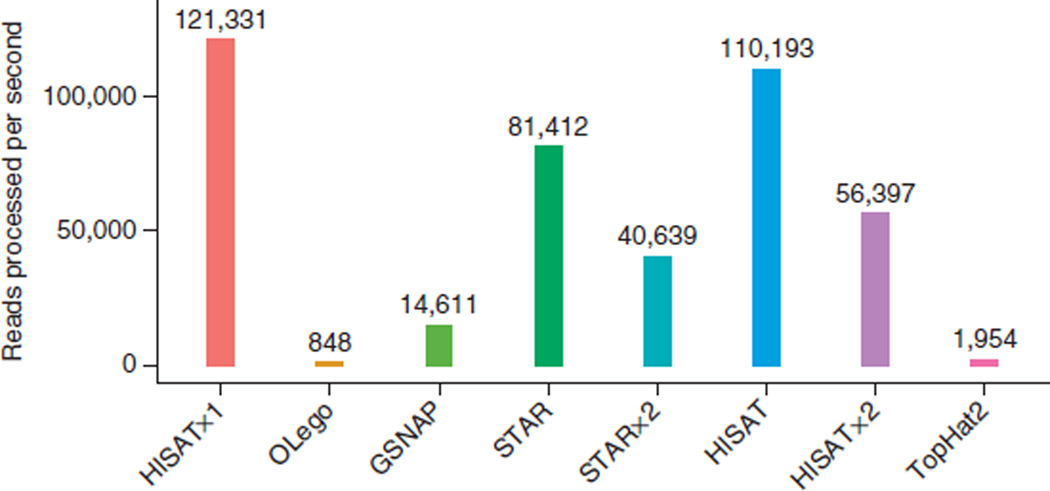
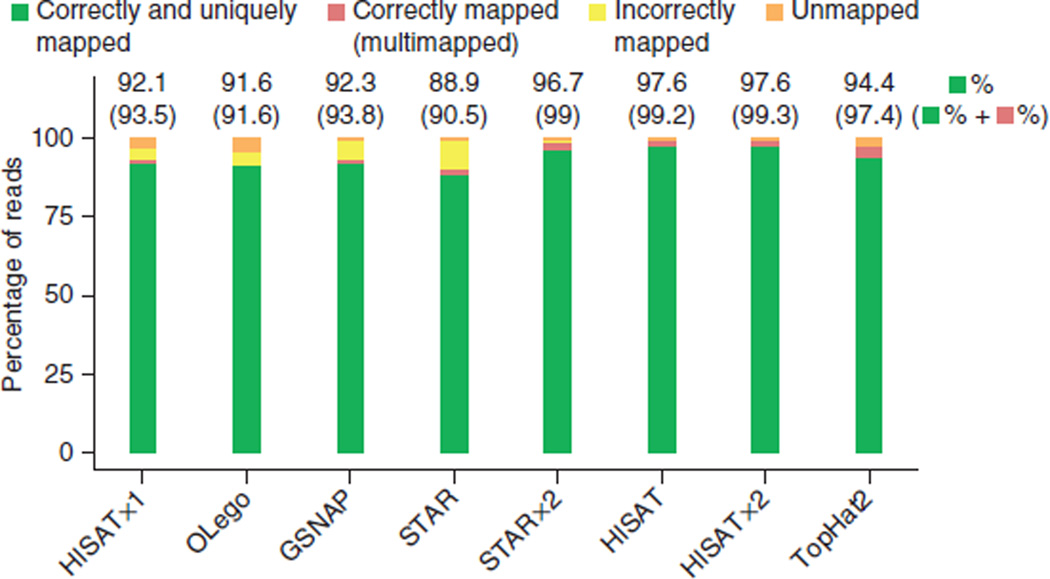
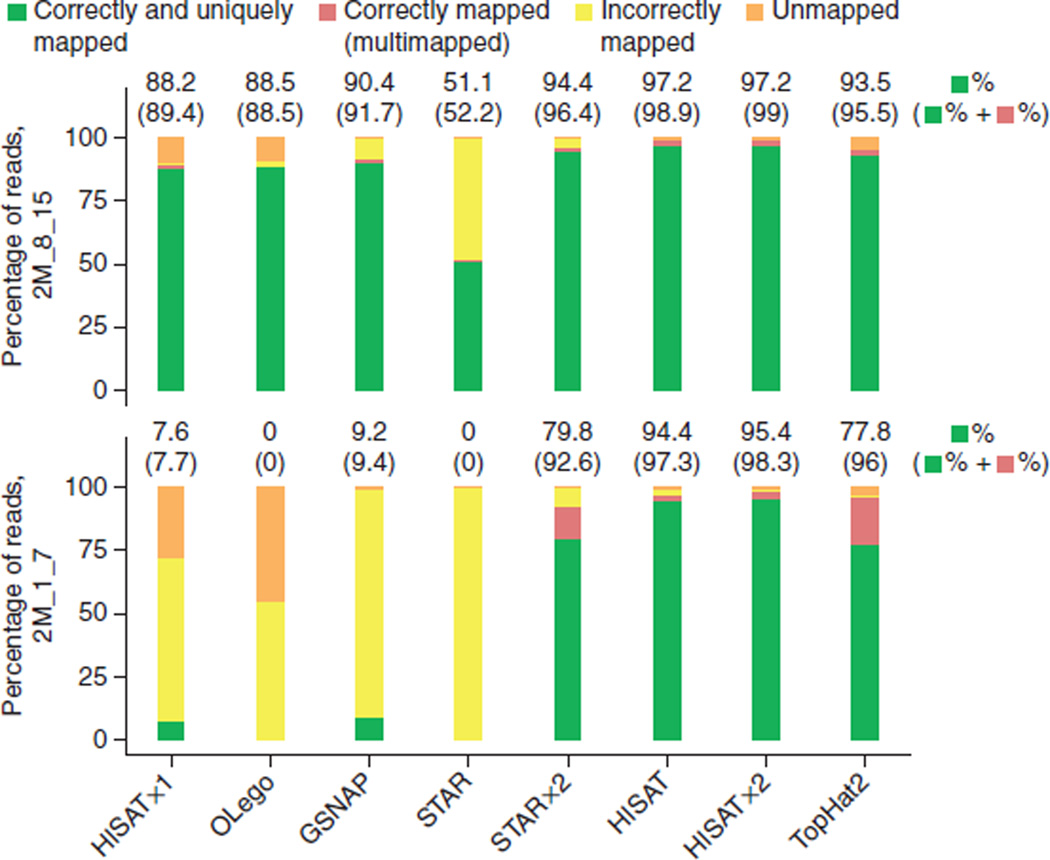
HISAT (hierarchical indexing for spliced alignment of transcripts) is a highly efficient system for aligning reads from RNA sequencing experiments. HISAT uses an indexing scheme based on the Burrows-Wheeler transform and the Ferragina-Manzini (FM) index, employing two types of indexes for alignment: a whole-genome FM index to anchor each alignment and numerous local FM indexes for very rapid extensions of these alignments. HISAT's hierarchical index for the human genome contains 48,000 local FM indexes, each representing a genomic region of ∼64,000 bp. Tests on real and simulated data sets showed that HISAT is the fastest system currently available, with equal or better accuracy than any other method. Despite its large number of indexes, HISAT requires only 4.3 gigabytes of memory. HISAT supports genomes of any size, including those larger than 4 billion bases.
Figures
{kind=link}
{kind=link}
{kind=link}
{kind=link}
References
-
- Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods. 2008;5:621–628. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases