Solving the problem of comparing whole bacterial genomes across different sequencing platforms
- PMID: 25110940
- PMCID: PMC4128722
- DOI: 10.1371/journal.pone.0104984
Solving the problem of comparing whole bacterial genomes across different sequencing platforms
Abstract
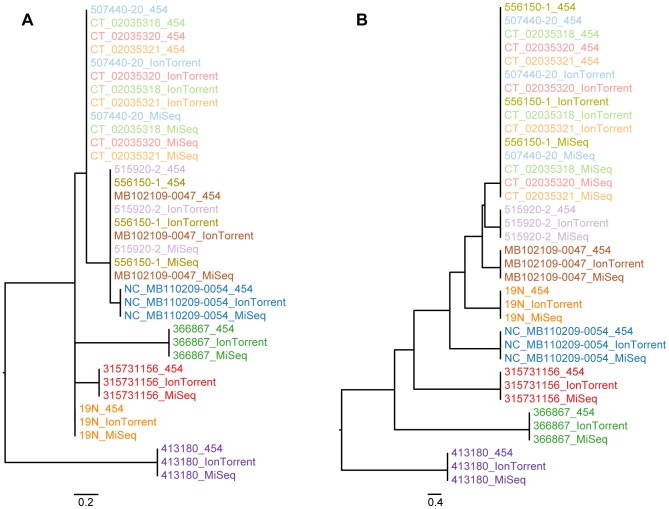
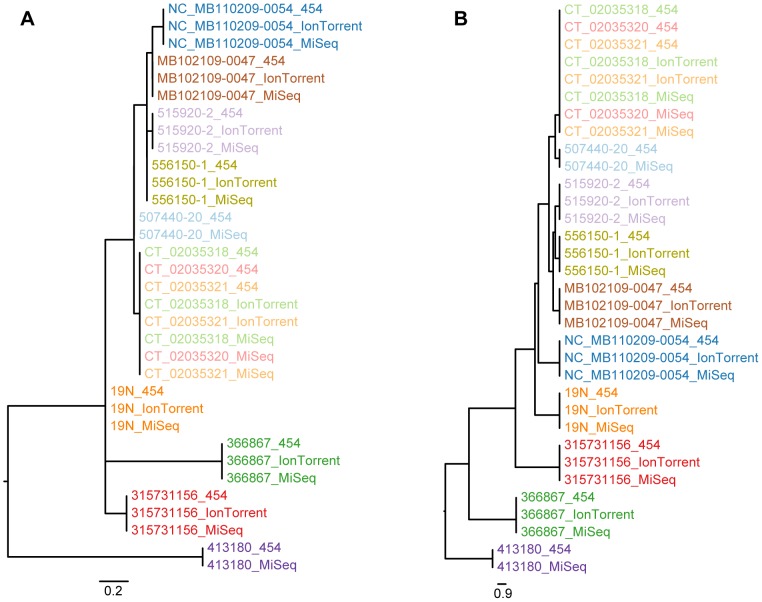
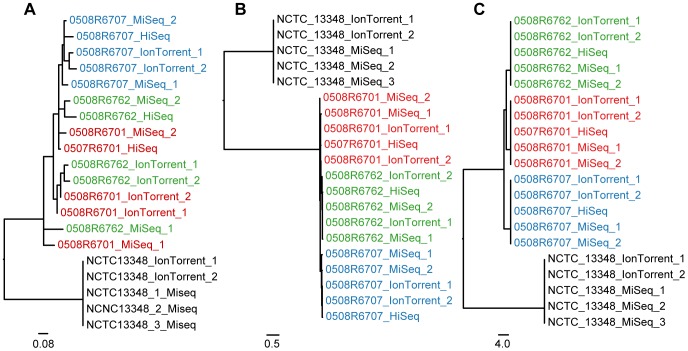
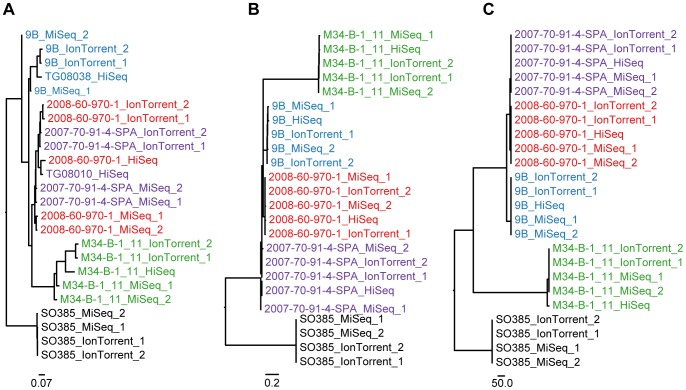
Whole genome sequencing (WGS) shows great potential for real-time monitoring and identification of infectious disease outbreaks. However, rapid and reliable comparison of data generated in multiple laboratories and using multiple technologies is essential. So far studies have focused on using one technology because each technology has a systematic bias making integration of data generated from different platforms difficult. We developed two different procedures for identifying variable sites and inferring phylogenies in WGS data across multiple platforms. The methods were evaluated on three bacterial data sets and sequenced on three different platforms (Illumina, 454, Ion Torrent). We show that the methods are able to overcome the systematic biases caused by the sequencers and infer the expected phylogenies. It is concluded that the cause of the success of these new procedures is due to a validation of all informative sites that are included in the analysis. The procedures are available as web tools.
Conflict of interest statement
Figures
{kind=link}
{kind=link}
{kind=link}
{kind=link}
References
-
- Aarestrup FM, Brown EW, Detter C, Gerner-Smidt P, Gilmour MW, et al. (2012) Integrating genome-based informatics to modernize global disease monitoring, information sharing, and response. Emerg Infect Dis 18: e1 Available: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3559169&tool=p... Accessed 27 February 2014.. - PMC - PubMed
-
- Didelot X, Bowden R, Wilson DJ, Peto TE, Crook DW (2012) Transforming clinical microbiology with bacterial genome sequencing. Nat Rev Genet 13: 601–612 Available: http://www.ncbi.nlm.nih.gov/pubmed/22868263 Accessed 20 February 2014.. - PMC - PubMed
-
- Köser CU, Ellington MJ, Cartwright EJP, Gillespie SH, Brown NM, et al. (2012) Routine use of microbial whole genome sequencing in diagnostic and public health microbiology. PLoS Pathog 8: e1002824 Available: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3410874&tool=p... Accessed 24 February 2014.. - PMC - PubMed
-
- Harris SR, Cartwright EJP, Török ME, Holden MTG, Brown NM, et al. (2013) Whole-genome sequencing for analysis of an outbreak of meticillin-resistant Staphylococcus aureus: a descriptive study. Lancet Infect Dis 13: 130–136 Available: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3556525&tool=p... Accessed 27 February 2014.. - PMC - PubMed
-
- Price L, Stegger M, Hasman H, Aziz M, Larsen J (2012) Staphylococcus aureus CC398: Host Adaptation and Emergence of Methicillin Resistance in Livestock. MBio 3: 1–6 Available: http://mbio.asm.org/content/3/1/e00305-11.short Accessed 25 May 2012.. - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources