selscan: an efficient multithreaded program to perform EHH-based scans for positive selection
- PMID: 25015648
- PMCID: PMC4166924
- DOI: 10.1093/molbev/msu211
selscan: an efficient multithreaded program to perform EHH-based scans for positive selection
Abstract
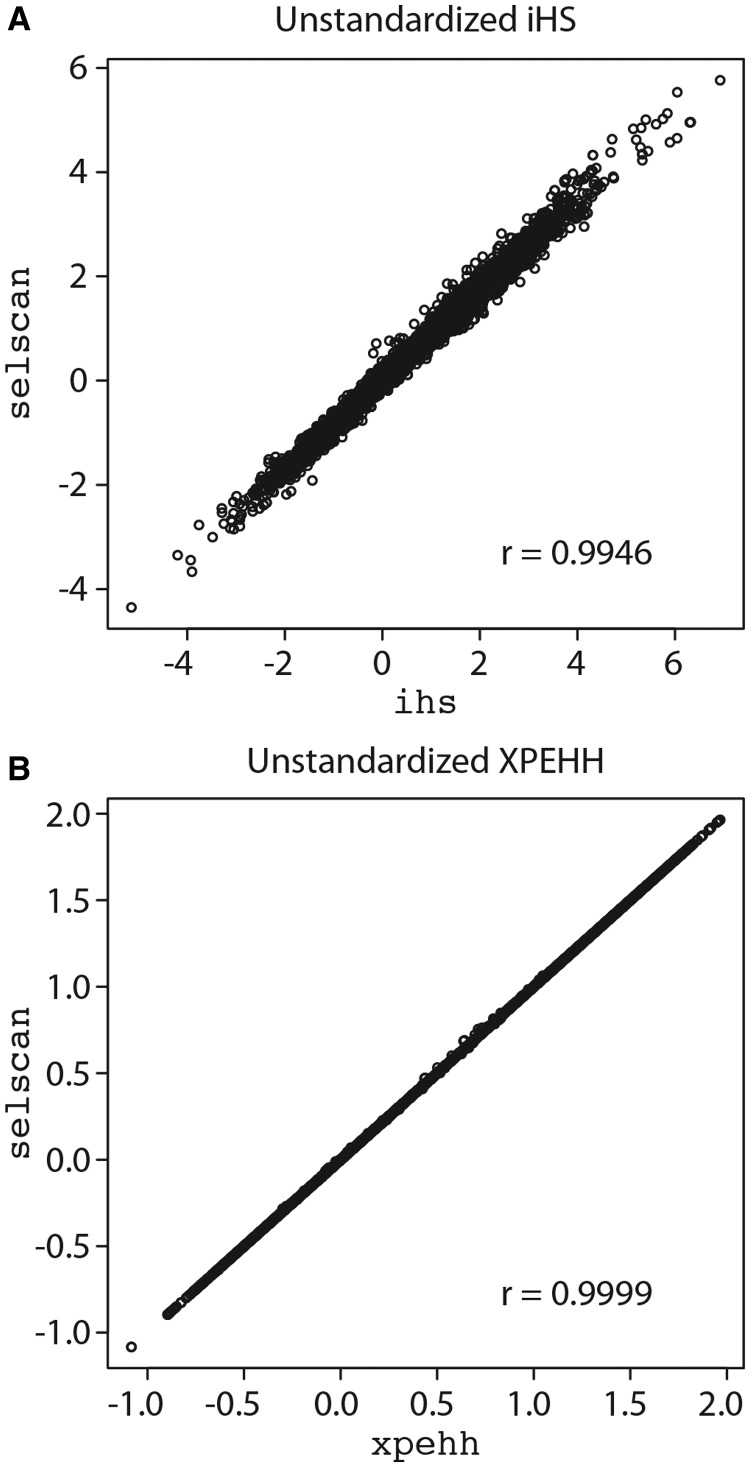
Haplotype-based scans to detect natural selection are useful to identify recent or ongoing positive selection in genomes. As both real and simulated genomic data sets grow larger, spanning thousands of samples and millions of markers, there is a need for a fast and efficient implementation of these scans for general use. Here, we present selscan, an efficient multithreaded application that implements Extended Haplotype Homozygosity (EHH), Integrated Haplotype Score (iHS), and Cross-population EHH (XPEHH). selscan accepts phased genotypes in multiple formats, including TPED, and performs extremely well on both simulated and real data and over an order of magnitude faster than existing available implementations. It calculates iHS on chromosome 22 (22,147 loci) across 204 CEU haplotypes in 353 s on one thread (33 s on 16 threads) and calculates XPEHH for the same data relative to 210 YRI haplotypes in 578 s on one thread (52 s on 16 threads). Source code and binaries (Windows, OSX, and Linux) are available at https://github.com/szpiech/selscan.
© The Author 2014. Published by Oxford University Press on behalf of the Society for Molecular Biology and Evolution.
Figures
{kind=link}
References
-
- Gautier M, Vitalis R. rehh: an R package to detect footprints of selection in genome-wide SNP data from haplotype structure. Bioinformatics. 2012;28(8):1176–1177. - PubMed
-
- Hudson RR. Generating samples under a wrightfisher neutral model of genetic variation. Bioinformatics. 2002;18(2):337–338. - PubMed
-
- Sabeti PC, Reich DE, Higgins JM, Levine HZP, Richter DJ, Schaffner SF, Gabriel SB, Platko JV, Patterson NJ, McDonald GJ, et al. Detecting recent positive selection in the human genome from haplotype structure. Nature. 2002;419(6909):832–837. - PubMed
Publication types
MeSH terms
Grants and funding
- 1R01HG007644/HG/NHGRI NIH HHS/United States
- UL1 RR024131/RR/NCRR NIH HHS/United States
- R01 HG007644/HG/NHGRI NIH HHS/United States
- P60MD006902/MD/NIMHD NIH HHS/United States
- UL1RR024131/RR/NCRR NIH HHS/United States
- 1R01HL117004-01/HL/NHLBI NIH HHS/United States
- UL1 TR000004/TR/NCATS NIH HHS/United States
- 1R21HG007233/HG/NHGRI NIH HHS/United States
- 1R21CA178706/CA/NCI NIH HHS/United States
- R21 CA178706/CA/NCI NIH HHS/United States
- R21 HG007233/HG/NHGRI NIH HHS/United States
- R01 HL117004/HL/NHLBI NIH HHS/United States
- P60 MD006902/MD/NIMHD NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials