Direct activation of β-cell KATP channels with a novel xanthine derivative
- PMID: 24646456
- PMCID: PMC4014665
- DOI: 10.1124/mol.114.091884
Direct activation of β-cell KATP channels with a novel xanthine derivative
Abstract
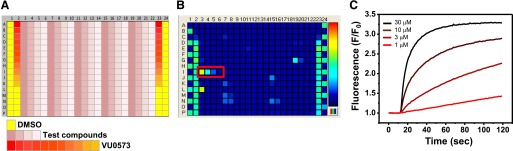
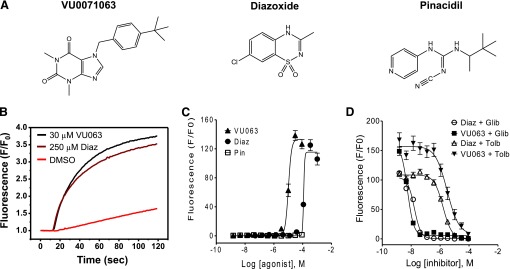
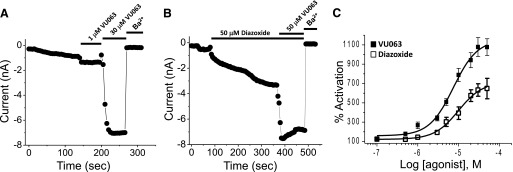
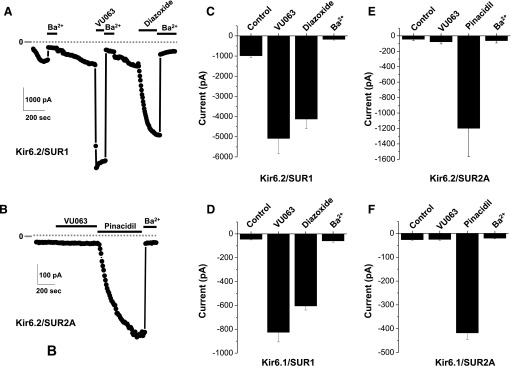
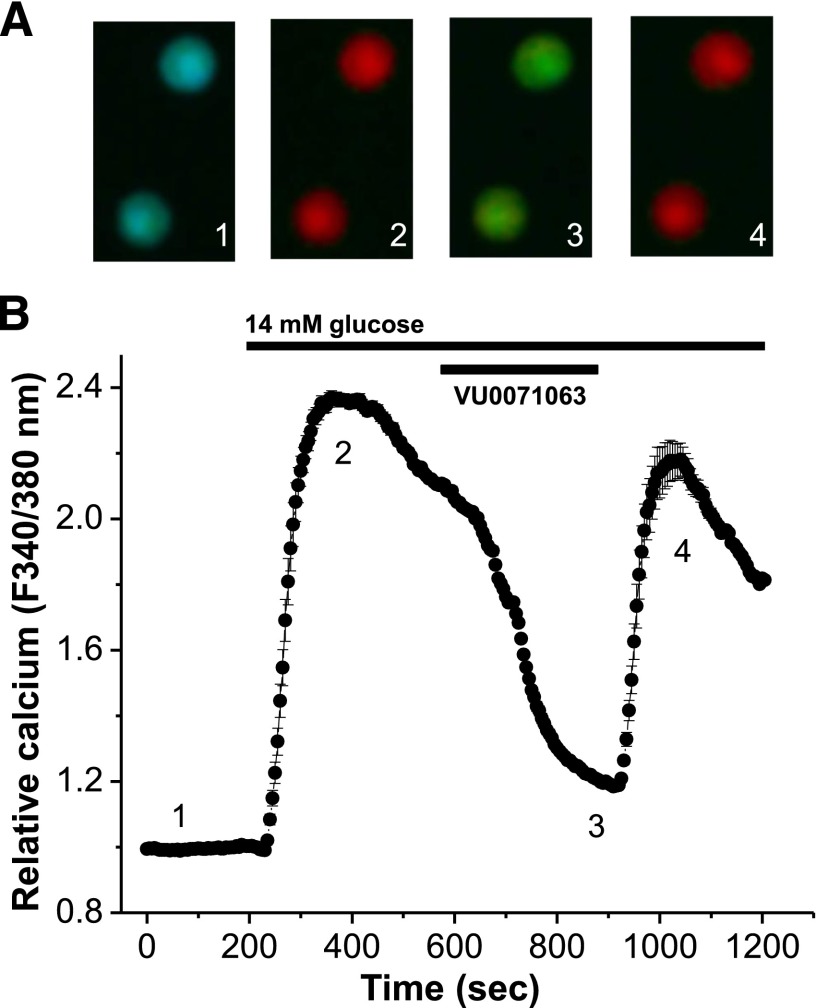
ATP-regulated potassium (KATP) channel complexes of inward rectifier potassium channel (Kir) 6.2 and sulfonylurea receptor (SUR) 1 critically regulate pancreatic islet β-cell membrane potential, calcium influx, and insulin secretion, and consequently, represent important drug targets for metabolic disorders of glucose homeostasis. The KATP channel opener diazoxide is used clinically to treat intractable hypoglycemia caused by excessive insulin secretion, but its use is limited by off-target effects due to lack of potency and selectivity. Some progress has been made in developing improved Kir6.2/SUR1 agonists from existing chemical scaffolds and compound screening, but there are surprisingly few distinct chemotypes that are specific for SUR1-containing KATP channels. Here we report the serendipitous discovery in a high-throughput screen of a novel activator of Kir6.2/SUR1: VU0071063 [7-(4-(tert-butyl)benzyl)-1,3-dimethyl-1H-purine-2,6(3H,7H)-dione]. The xanthine derivative rapidly and dose-dependently activates Kir6.2/SUR1 with a half-effective concentration (EC50) of approximately 7 μM, is more efficacious than diazoxide at low micromolar concentrations, directly activates the channel in excised membrane patches, and is selective for SUR1- over SUR2A-containing Kir6.1 or Kir6.2 channels, as well as Kir2.1, Kir2.2, Kir2.3, Kir3.1/3.2, and voltage-gated potassium channel 2.1. Finally, we show that VU0071063 activates native Kir6.2/SUR1 channels, thereby inhibiting glucose-stimulated calcium entry in isolated mouse pancreatic β cells. VU0071063 represents a novel tool/compound for investigating β-cell physiology, KATP channel gating, and a new chemical scaffold for developing improved activators with medicinal chemistry.
Figures
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
References
-
- Alemzadeh R, Fledelius C, Bodvarsdottir T, Sturis J. (2004) Attenuation of hyperinsulinemia by NN414, a SUR1/Kir6.2 selective K-adenosine triphosphate channel opener, improves glucose tolerance and lipid profile in obese Zucker rats. Metabolism 53:441–447 - PubMed
-
- Ashcroft FM. (1988) Adenosine 5′-triphosphate-sensitive potassium channels. Annu Rev Neurosci 11:97–118 - PubMed
-
- Ashcroft FM. (2007) The Walter B. Cannon Physiology in Perspective Lecture, 2007. ATP-sensitive K+ channels and disease: from molecule to malady. Am J Physiol Endocrinol Metab 293:E880–E889 - PubMed
-
- Carr RD, Brand CL, Bodvarsdottir TB, Hansen JB, Sturis J. (2003) NN414, a SUR1/Kir6.2-selective potassium channel opener, reduces blood glucose and improves glucose tolerance in the VDF Zucker rat. Diabetes 52:2513–2518 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources