Kinannote, a computer program to identify and classify members of the eukaryotic protein kinase superfamily
- PMID: 23904509
- PMCID: PMC3777111
- DOI: 10.1093/bioinformatics/btt419
Kinannote, a computer program to identify and classify members of the eukaryotic protein kinase superfamily
Abstract
Motivation: Kinases of the eukaryotic protein kinase superfamily are key regulators of most aspects eukaryotic cellular behavior and have provided several drug targets including kinases dysregulated in cancers. The rapid increase in the number of genomic sequences has created an acute need to identify and classify members of this important class of enzymes efficiently and accurately.
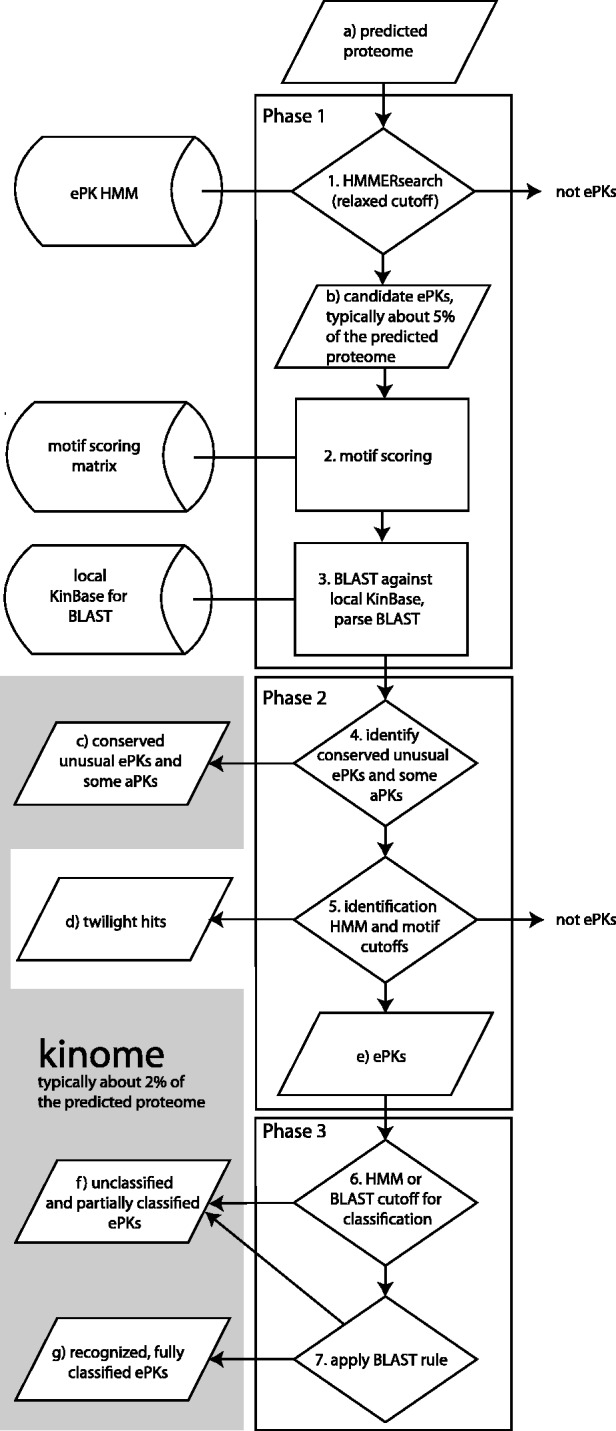
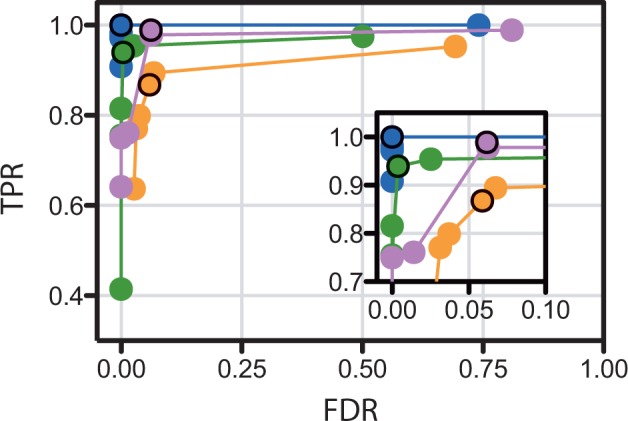
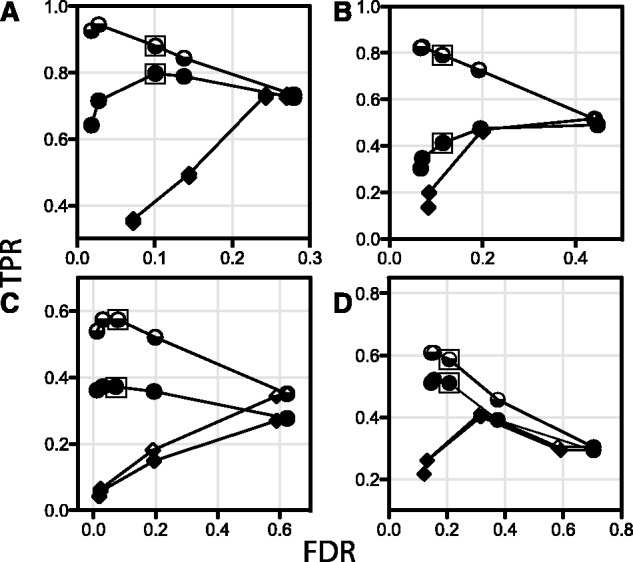
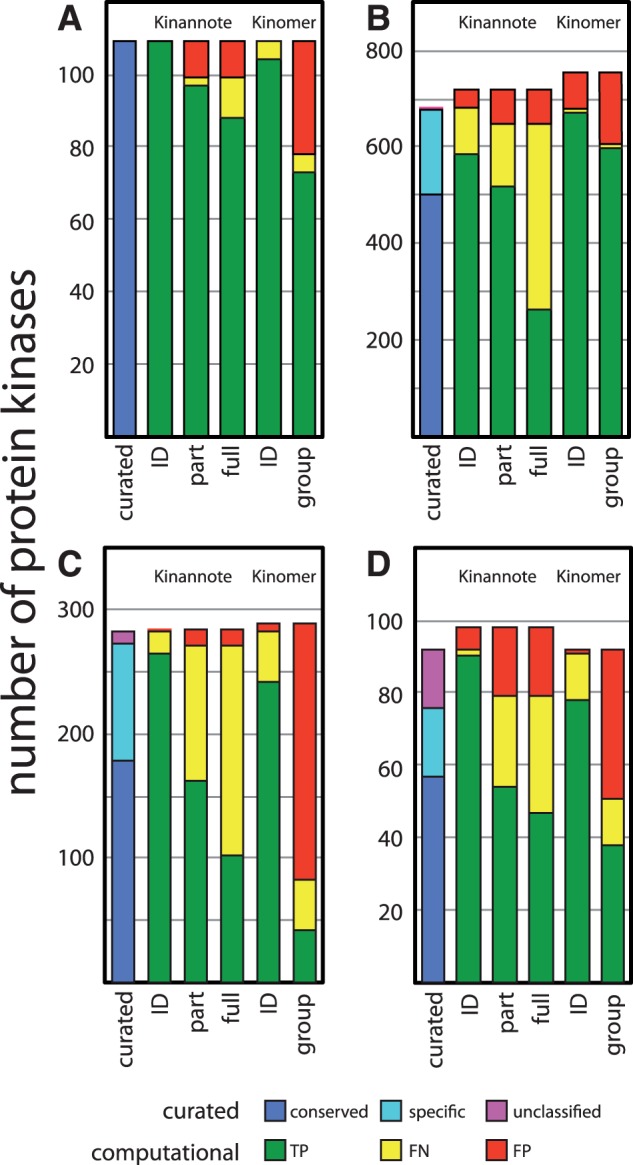
Results: Kinannote produces a draft kinome and comparative analyses for a predicted proteome using a single line command, and it is currently the only tool that automatically classifies protein kinases using the controlled vocabulary of Hanks and Hunter [Hanks and Hunter (1995)]. A hidden Markov model in combination with a position-specific scoring matrix is used by Kinannote to identify kinases, which are subsequently classified using a BLAST comparison with a local version of KinBase, the curated protein kinase dataset from www.kinase.com. Kinannote was tested on the predicted proteomes from four divergent species. The average sensitivity and precision for kinome retrieval from the test species are 94.4 and 96.8%. The ability of Kinannote to classify identified kinases was also evaluated, and the average sensitivity and precision for full classification of conserved kinases are 71.5 and 82.5%, respectively. Kinannote has had a significant impact on eukaryotic genome annotation, providing protein kinase annotations for 36 genomes made public by the Broad Institute in the period spanning 2009 to the present.
Availability: Kinannote is freely available at http://sourceforge.net/projects/kinannote.
Figures
{kind=link}
{kind=link}
{kind=link}
{kind=link}
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials