The branch-site test of positive selection is surprisingly robust but lacks power under synonymous substitution saturation and variation in GC
- PMID: 23558341
- PMCID: PMC3684852
- DOI: 10.1093/molbev/mst062
The branch-site test of positive selection is surprisingly robust but lacks power under synonymous substitution saturation and variation in GC
Abstract
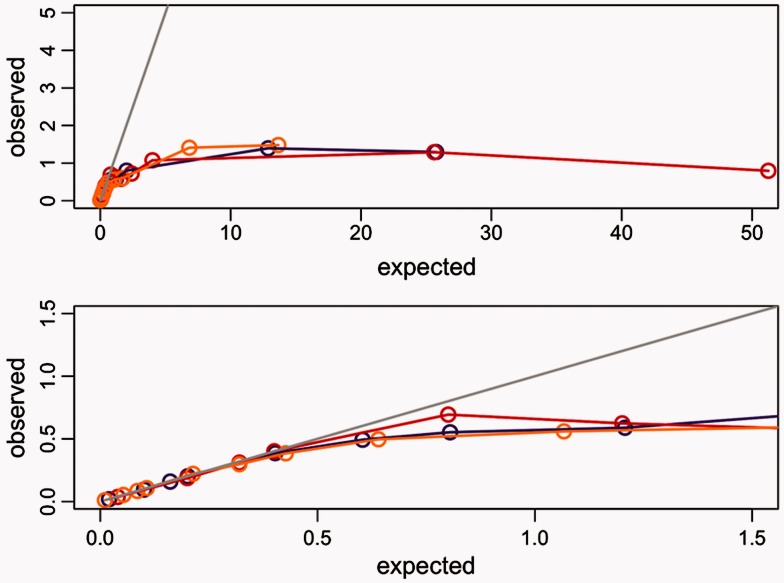
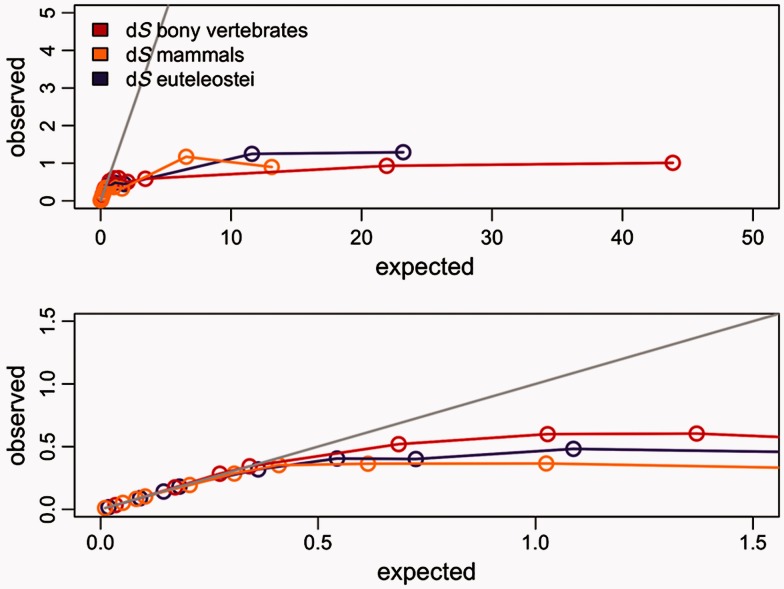
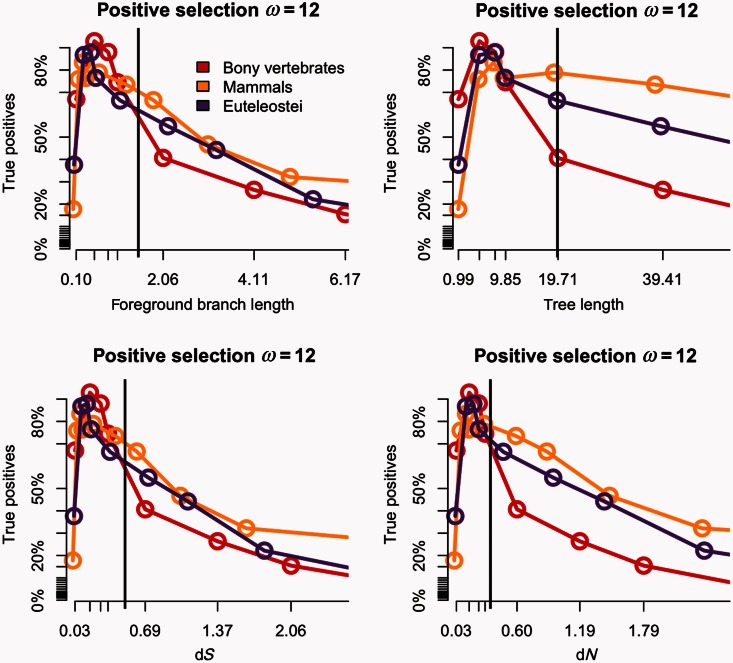
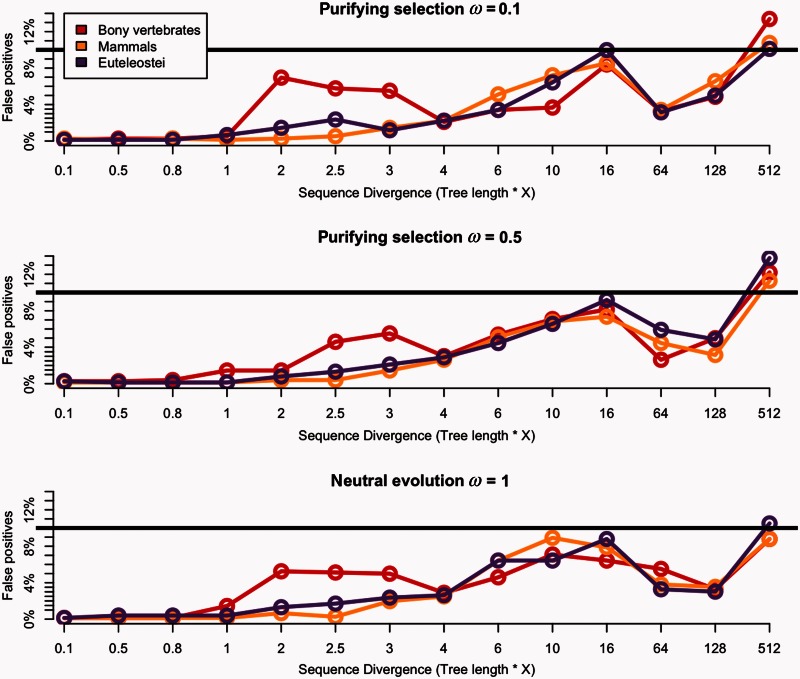
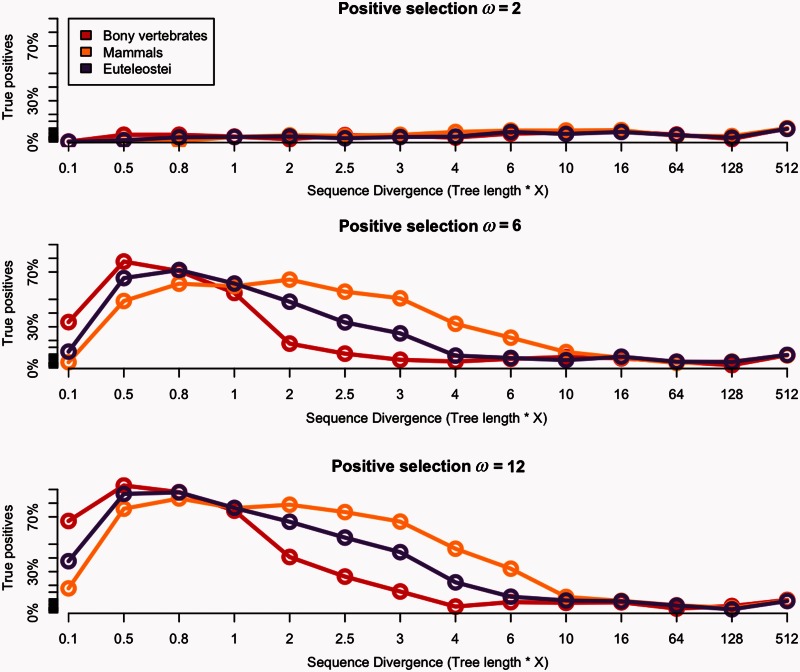
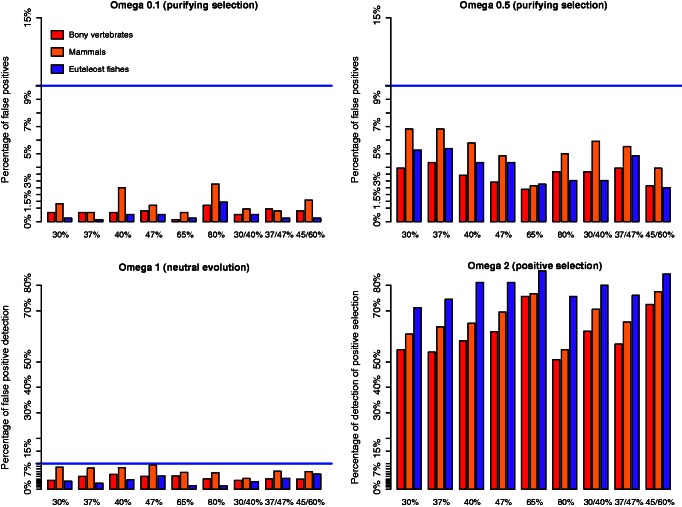
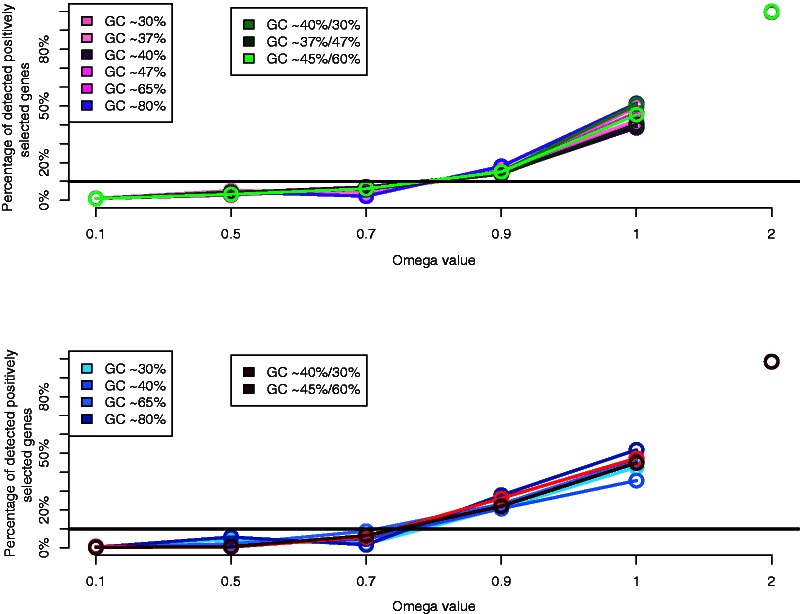
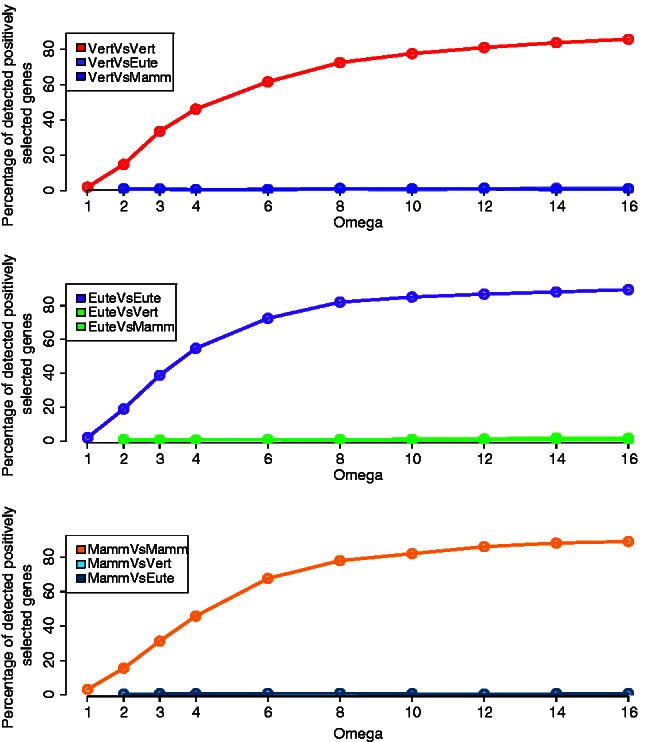
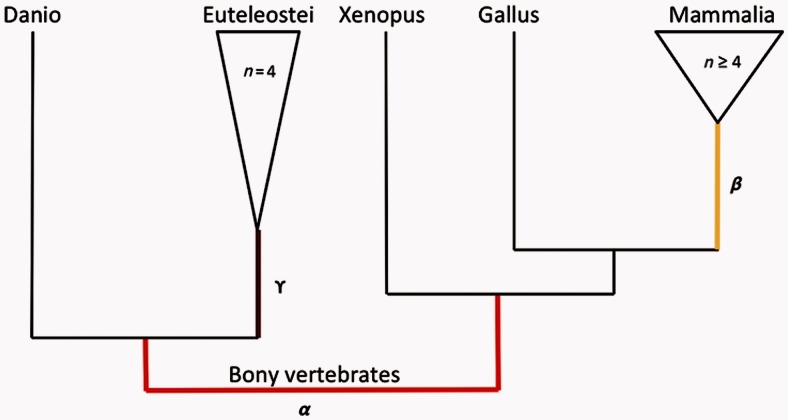
Positive selection is widely estimated from protein coding sequence alignments by the nonsynonymous-to-synonymous ratio ω. Increasingly elaborate codon models are used in a likelihood framework for this estimation. Although there is widespread concern about the robustness of the estimation of the ω ratio, more efforts are needed to estimate this robustness, especially in the context of complex models. Here, we focused on the branch-site codon model. We investigated its robustness on a large set of simulated data. First, we investigated the impact of sequence divergence. We found evidence of underestimation of the synonymous substitution rate for values as small as 0.5, with a slight increase in false positives for the branch-site test. When dS increases further, underestimation of dS is worse, but false positives decrease. Interestingly, the detection of true positives follows a similar distribution, with a maximum for intermediary values of dS. Thus, high dS is more of a concern for a loss of power (false negatives) than for false positives of the test. Second, we investigated the impact of GC content. We showed that there is no significant difference of false positives between high GC (up to ∼80%) and low GC (∼30%) genes. Moreover, neither shifts of GC content on a specific branch nor major shifts in GC along the gene sequence generate many false positives. Our results confirm that the branch-site is a very conservative test.
Keywords: adaptive evolution; base composition; codon model.
Figures
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
References
-
- Anisimova M, Bielawski JP, Yang Z. Accuracy and power of the likelihood ratio test in detecting adaptive molecular evolution. Mol Biol Evol. 2001;18:1585–1592. - PubMed
-
- Anisimova M, Bielawski JP, Yang Z. Accuracy and power of bayes prediction of amino acid sites under positive selection. Mol Biol Evol. 2002;19:950–958. - PubMed
-
- Anisimova M, Kosiol C. Investigating protein-coding sequence evolution with probabilistic codon substitution models. Mol Biol Evol. 2009;26:255–271. - PubMed
-
- Anisimova M, Yang Z. Multiple hypothesis testing to detect lineages under positive selection that affects only a few sites. Mol Biol Evol. 2007;24:1219–1228. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous