A systematically improved high quality genome and transcriptome of the human blood fluke Schistosoma mansoni
- PMID: 22253936
- PMCID: PMC3254664
- DOI: 10.1371/journal.pntd.0001455
A systematically improved high quality genome and transcriptome of the human blood fluke Schistosoma mansoni
Abstract
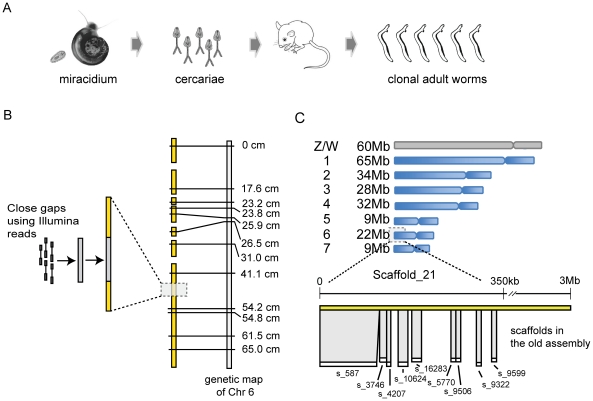
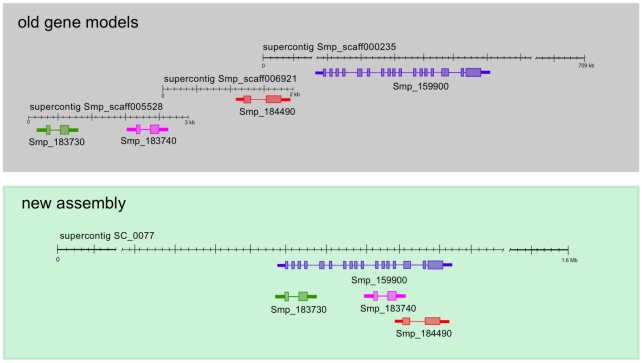
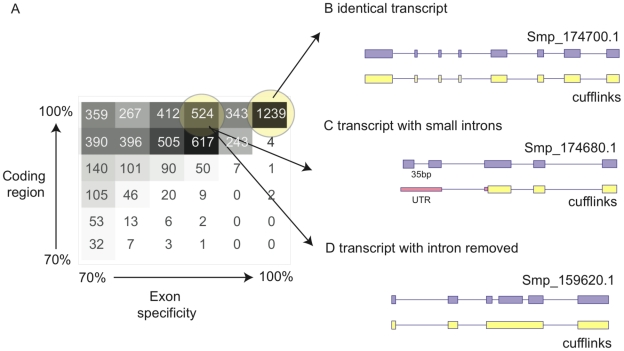
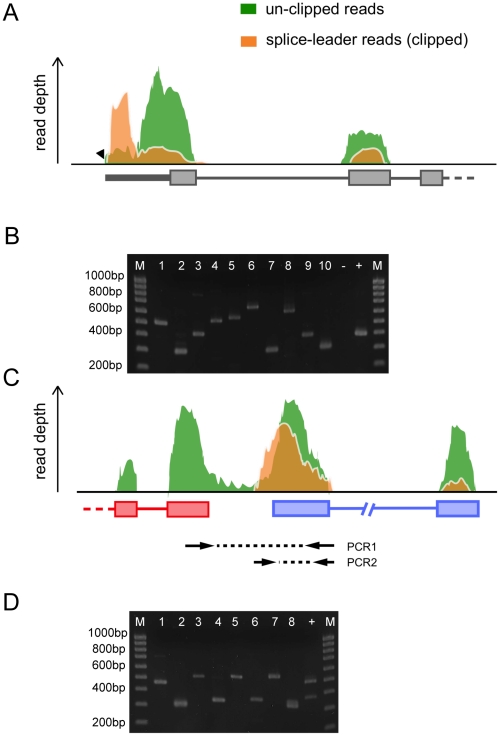
Schistosomiasis is one of the most prevalent parasitic diseases, affecting millions of people in developing countries. Amongst the human-infective species, Schistosoma mansoni is also the most commonly used in the laboratory and here we present the systematic improvement of its draft genome. We used Sanger capillary and deep-coverage Illumina sequencing from clonal worms to upgrade the highly fragmented draft 380 Mb genome to one with only 885 scaffolds and more than 81% of the bases organised into chromosomes. We have also used transcriptome sequencing (RNA-seq) from four time points in the parasite's life cycle to refine gene predictions and profile their expression. More than 45% of predicted genes have been extensively modified and the total number has been reduced from 11,807 to 10,852. Using the new version of the genome, we identified trans-splicing events occurring in at least 11% of genes and identified clear cases where it is used to resolve polycistronic transcripts. We have produced a high-resolution map of temporal changes in expression for 9,535 genes, covering an unprecedented dynamic range for this organism. All of these data have been consolidated into a searchable format within the GeneDB (www.genedb.org) and SchistoDB (www.schistodb.net) databases. With further transcriptional profiling and genome sequencing increasingly accessible, the upgraded genome will form a fundamental dataset to underpin further advances in schistosome research.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
References
-
- Steinmann P, Keiser J, Bos R, Tanner M, Utzinger J. Schistosomiasis and water resources development: systematic review, meta-analysis, and estimates of people at risk. Lancet Infect Dis. 2006;6:411–425. - PubMed
-
- Cook GC, Zumla A, Manson PSTd. Manson's tropical diseases. Edinburgh: Saunders; 2003. [1812] p. of plates p. xiii, 1847.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous