Multilocus sequence typing of total-genome-sequenced bacteria
- PMID: 22238442
- PMCID: PMC3318499
- DOI: 10.1128/JCM.06094-11
Multilocus sequence typing of total-genome-sequenced bacteria
Abstract
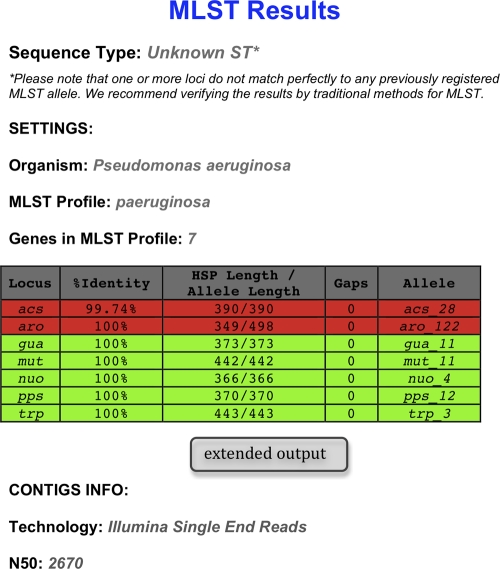
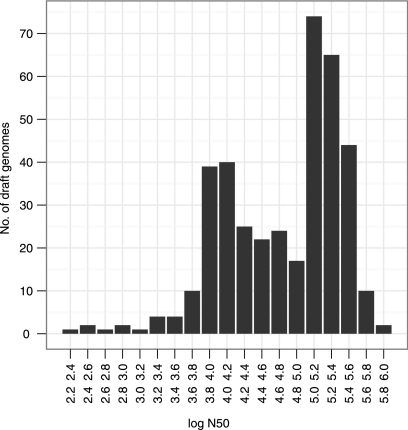
Accurate strain identification is essential for anyone working with bacteria. For many species, multilocus sequence typing (MLST) is considered the "gold standard" of typing, but it is traditionally performed in an expensive and time-consuming manner. As the costs of whole-genome sequencing (WGS) continue to decline, it becomes increasingly available to scientists and routine diagnostic laboratories. Currently, the cost is below that of traditional MLST. The new challenges will be how to extract the relevant information from the large amount of data so as to allow for comparison over time and between laboratories. Ideally, this information should also allow for comparison to historical data. We developed a Web-based method for MLST of 66 bacterial species based on WGS data. As input, the method uses short sequence reads from four sequencing platforms or preassembled genomes. Updates from the MLST databases are downloaded monthly, and the best-matching MLST alleles of the specified MLST scheme are found using a BLAST-based ranking method. The sequence type is then determined by the combination of alleles identified. The method was tested on preassembled genomes from 336 isolates covering 56 MLST schemes, on short sequence reads from 387 isolates covering 10 schemes, and on a small test set of short sequence reads from 29 isolates for which the sequence type had been determined by traditional methods. The method presented here enables investigators to determine the sequence types of their isolates on the basis of WGS data. This method is publicly available at www.cbs.dtu.dk/services/MLST.
Figures
{kind=link}
{kind=link}
References
-
- Bielaszewska M, et al. 2011. Characterisation of the Escherichia coli strain associated with an outbreak of haemolytic uraemic syndrome in Germany, 2011: a microbiological study. Lancet Infect. Dis. 11:671–676 - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials