Post-acquisition ETD spectral processing for increased peptide identifications
- PMID: 19362853
- PMCID: PMC2716440
- DOI: 10.1016/j.jasms.200903006
Post-acquisition ETD spectral processing for increased peptide identifications
Abstract
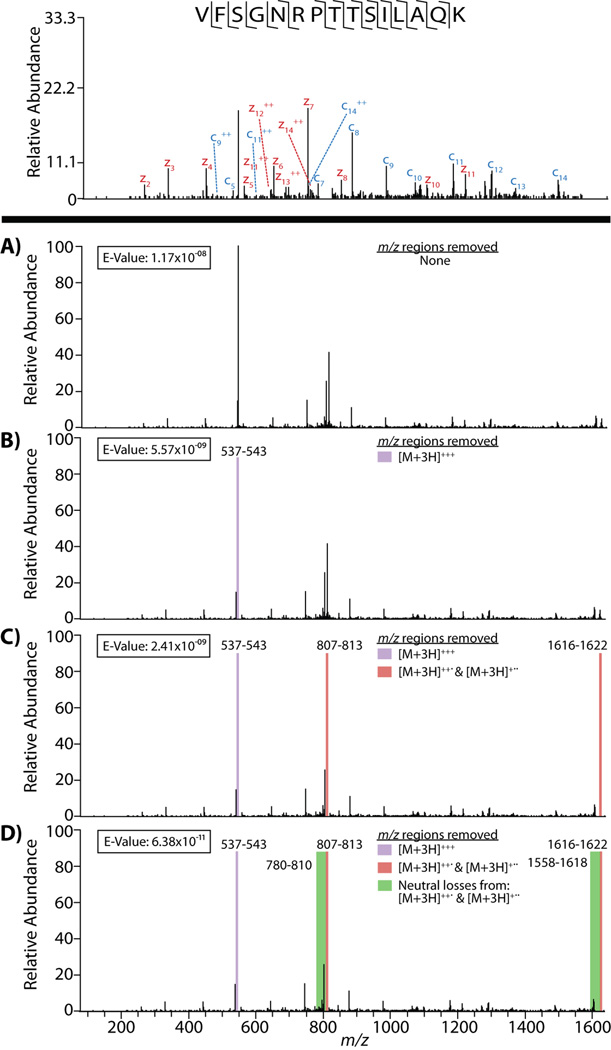
Tandem mass spectra (MS/MS) produced using electron transfer dissociation (ETD) differ from those derived from collision-activated dissociation (CAD) in several important ways. Foremost, the predominant fragment ion series are different: c- and z(*)-type ions are favored in ETD spectra while b- and y-type ions comprise the bulk of the fragments in CAD spectra. Additionally, ETD spectra possess charge-reduced precursors and unique neutral losses. Most database search algorithms were designed to analyze CAD spectra, and have only recently been adapted to accommodate c- and z(*)-type ions; therefore, inclusion of these additional spectral features can hinder identification, leading to lower confidence scores and decreased sensitivity. Because of this, it is important to pre-process spectral data before submission to a database search to remove those features that cause complications. Here, we demonstrate the effects of removing these features on the number of unique peptide identifications at a 1% false discovery rate (FDR) using the open mass spectrometry search algorithm (OMSSA). When analyzing two biologic replicates of a yeast protein extract in three total analyses, the number of unique identifications with a approximately 1% FDR increased from 4611 to 5931 upon spectral pre-processing--an increase of approximately 28.6%. We outline the most effective pre-processing methods, and provide free software containing these algorithms.
Figures
{kind=link}
{kind=link}
References
-
- Zubarev RA, Kelleher NL, McLafferty FW. Electron capture dissociation of multiply charged protein cations. A nonergodic process. Journal of the American Chemical Society. 1998;120(13):3265–3266.
-
- Chi A, Huttenhower C, Geer LY, Coon JJ, Syka JEP, Bai DL, Shabanowitz J, Burke DJ, Troyanskaya OG, Hunt DF. Analysis of phosphorylation sites on proteins from Saccharomyces cerevisiae by electron transfer dissociation (ETD) mass spectrometry. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(7):2193–2198. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous