Sex and virulence in Escherichia coli: an evolutionary perspective
- PMID: 16689791
- PMCID: PMC1557465
- DOI: 10.1111/j.1365-2958.2006.05172.x
Sex and virulence in Escherichia coli: an evolutionary perspective
Abstract
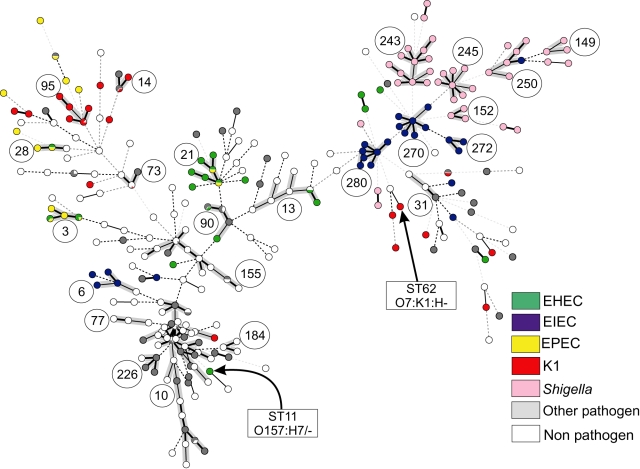
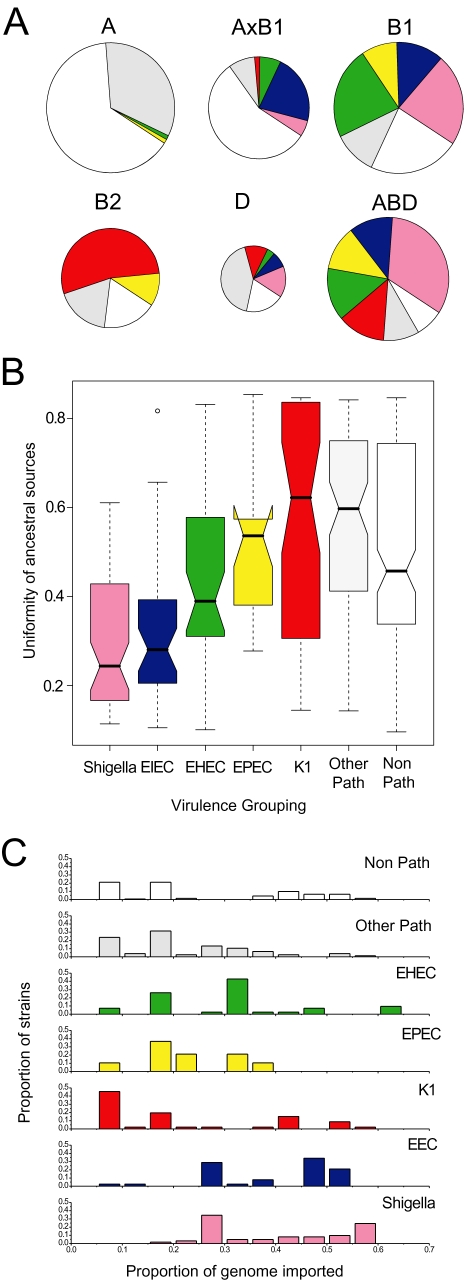
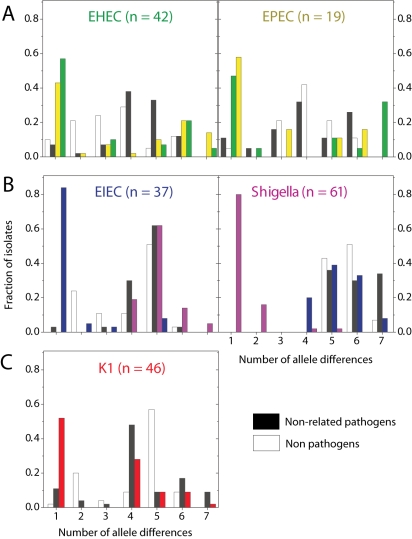
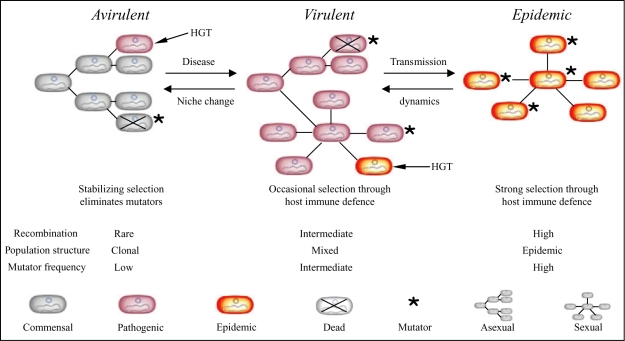
Pathogenic Escherichia coli cause over 160 million cases of dysentery and one million deaths per year, whereas non-pathogenic E. coli constitute part of the normal intestinal flora of healthy mammals and birds. The evolutionary pathways underlying this dichotomy in bacterial lifestyle were investigated by multilocus sequence typing of a global collection of isolates. Specific pathogen types [enterohaemorrhagic E. coli, enteropathogenic E. coli, enteroinvasive E. coli, K1 and Shigella] have arisen independently and repeatedly in several lineages, whereas other lineages contain only few pathogens. Rates of evolution have accelerated in pathogenic lineages, culminating in highly virulent organisms whose genomic contents are altered frequently by increased rates of homologous recombination; thus, the evolution of virulence is linked to bacterial sex. This long-term pattern of evolution was observed in genes distributed throughout the genome, and thereby is the likely result of episodic selection for strains that can escape the host immune response.
Figures
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources