MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform
- PMID: 12136088
- PMCID: PMC135756
- DOI: 10.1093/nar/gkf436
MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform
Abstract
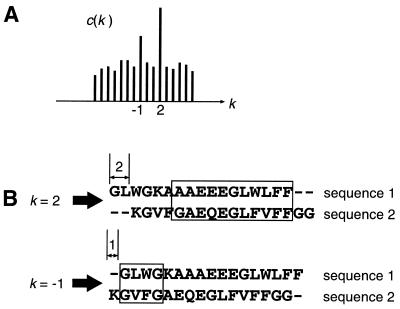
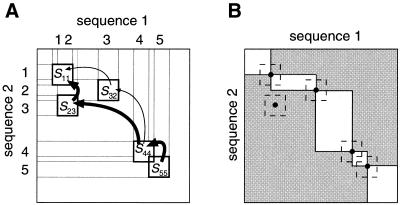
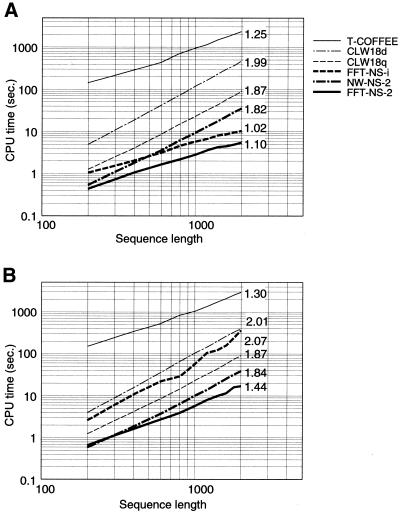
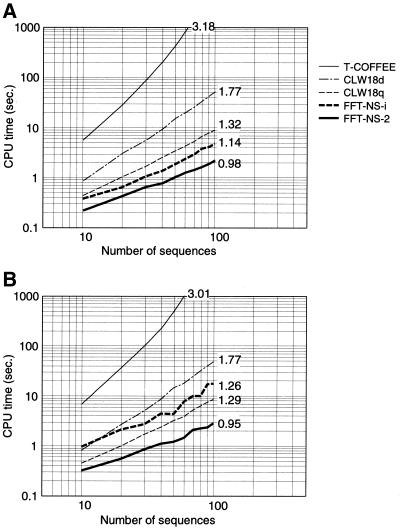
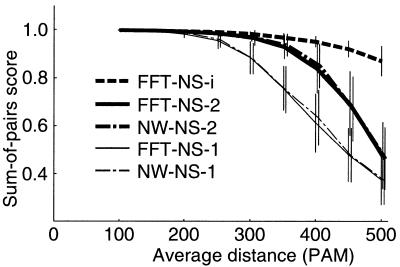
A multiple sequence alignment program, MAFFT, has been developed. The CPU time is drastically reduced as compared with existing methods. MAFFT includes two novel techniques. (i) Homo logous regions are rapidly identified by the fast Fourier transform (FFT), in which an amino acid sequence is converted to a sequence composed of volume and polarity values of each amino acid residue. (ii) We propose a simplified scoring system that performs well for reducing CPU time and increasing the accuracy of alignments even for sequences having large insertions or extensions as well as distantly related sequences of similar length. Two different heuristics, the progressive method (FFT-NS-2) and the iterative refinement method (FFT-NS-i), are implemented in MAFFT. The performances of FFT-NS-2 and FFT-NS-i were compared with other methods by computer simulations and benchmark tests; the CPU time of FFT-NS-2 is drastically reduced as compared with CLUSTALW with comparable accuracy. FFT-NS-i is over 100 times faster than T-COFFEE, when the number of input sequences exceeds 60, without sacrificing the accuracy.
Figures
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
References
-
- Needleman S.B., and Wunsch,C.D. (1970) A general method applicable to the search for similarities in the amino acid sequence of two proteins. J. Mol. Biol., 48, 443–453. - PubMed
-
- Sankoff D., and Cedergren,R.J. (1983) Simultaneous comparison of three or more sequences related by a tree. In Sankoff,D. and Kruskal,J.B. (eds), Time Warps, String Edits and Macromolecules: The Theory and Practice of Sequence Comparison. Addison-Wesley, London, UK, pp. 253–264.
-
- Feng D.F., and Doolittle,R.F. (1987) Progressive sequence alignment as a prerequisite to correct phylogenetic trees. J. Mol. Evol., 25, 351–360. - PubMed
-
- Barton G.J., and Sternberg,M.J. (1987) A strategy for the rapid multiple alignment of protein sequences. Confidence levels from tertiary structure comparisons. J. Mol. Biol., 198, 327–337. - PubMed
-
- Berger M.P., and Munson,P.J. (1991) A novel randomized iterative strategy for aligning multiple protein sequences. Comput. Appl. Biosci., 7, 479–484. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous