Extensive Copy Number Variation Explains Genome Size Variation in the Unicellular Zygnematophycean Alga, Closterium peracerosum-strigosum-littorale Complex
- PMID: 37348049
- PMCID: PMC10407611
- DOI: 10.1093/gbe/evad115
Extensive Copy Number Variation Explains Genome Size Variation in the Unicellular Zygnematophycean Alga, Closterium peracerosum-strigosum-littorale Complex
Abstract
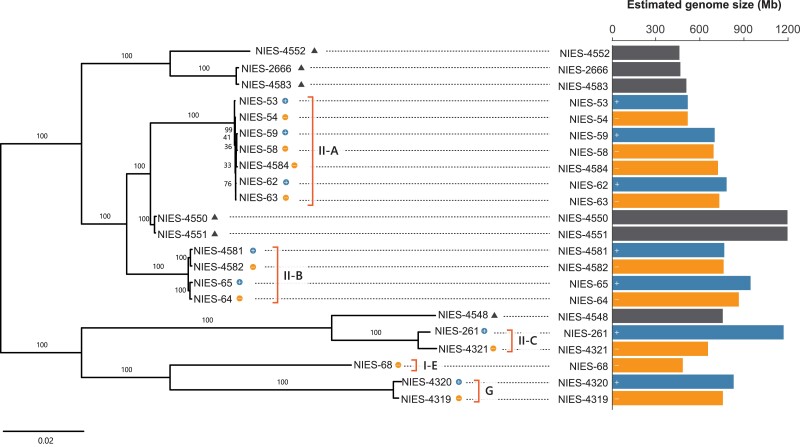
Genome sizes are known to vary within and among closely related species, but the knowledge about genomic factors contributing to the variation and their impacts on gene functions is limited to only a small number of species. This study identified a more than 2-fold heritable genome size variation among the unicellular Zygnematophycean alga, Closterium peracerosum-strigosum-littorale (C. psl.) complex, based on short-read sequencing analysis of 22 natural strains and F1 segregation analysis. Six de novo assembled genomes revealed that genome size variation is largely attributable to genome-wide copy number variation (CNV) among strains rather than mating type-linked genomic regions or specific repeat sequences such as rDNA. Notably, about 30% of genes showed CNV even between strains that can mate with each other. Transcriptome and gene ontology analysis demonstrated that CNV is distributed nonrandomly in terms of gene functions, such that CNV was more often observed in the gene set with stage-specific expression. Furthermore, in about 30% of these genes with CNV, the expression level does not increase proportionally with the gene copy number, suggesting presence of dosage compensation, which was overrepresented in genes involved in basic biological functions, such as translation. Nonrandom patterns in gene duplications and corresponding expression changes in terms of gene functions may contribute to maintaining the high level of CNV associated with extensive genome size variation in the C. psl. complex, despite its possible detrimental effects.
Keywords: copy number variation; dosage compensation; gene duplication; green algae.
© The Author(s) 2023. Published by Oxford University Press on behalf of Society for Molecular Biology and Evolution.
Figures
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
References
-
- Ågren JA, Wright SI. 2011. Co-evolution between transposable elements and their hosts: a major factor in genome size evolution? Chromosome Res. 19:777–786. - PubMed
-
- Alexa A, Rahnenführer J, Lengauer T. 2006. Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics 22:1600–1607. - PubMed
-
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J Mol Biol. 215:403–410. - PubMed
-
- Ameijeiras-Alonso J, Crujeiras RM, Rodriguez-Casal A. 2021. Multimode: an R package for mode assessment. J Stat Softw. 97:1–32.
-
- Biémont C. 2008. Genome size evolution: within-species variation in genome size. Heredity (Edinb). 101:297–298. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Research Materials