Precise phylogenetic analysis of microbial isolates and genomes from metagenomes using PhyloPhlAn 3.0
- PMID: 32427907
- PMCID: PMC7237447
- DOI: 10.1038/s41467-020-16366-7
Precise phylogenetic analysis of microbial isolates and genomes from metagenomes using PhyloPhlAn 3.0
Abstract
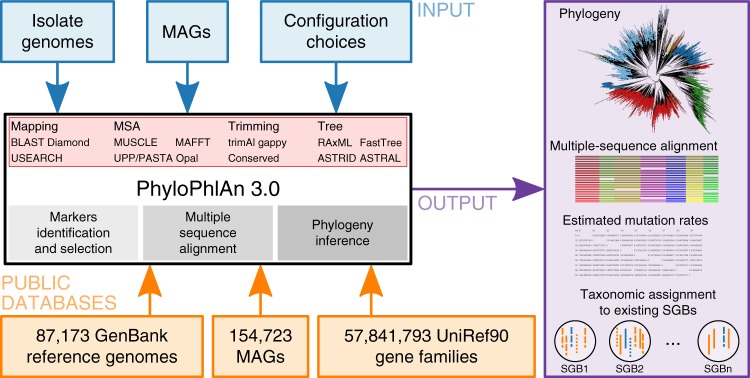
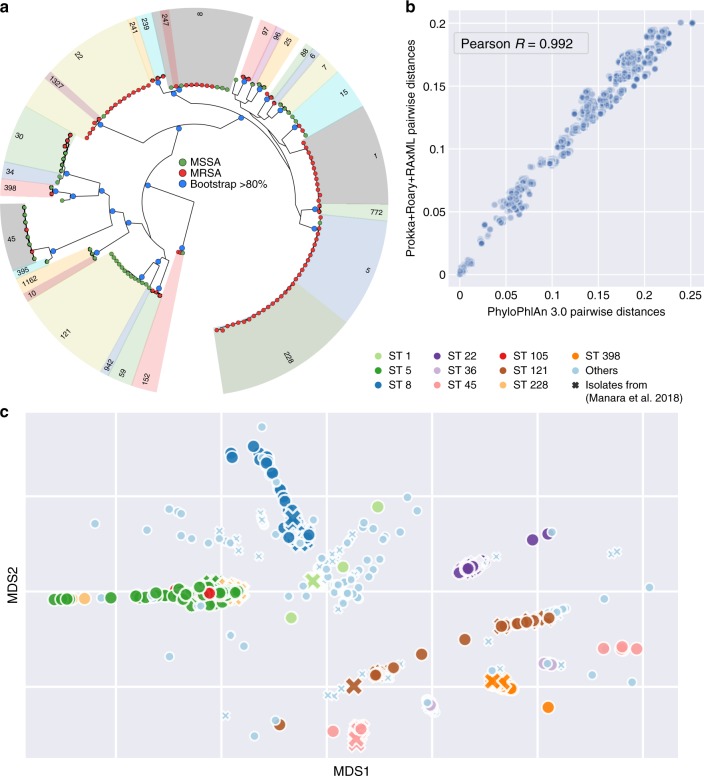
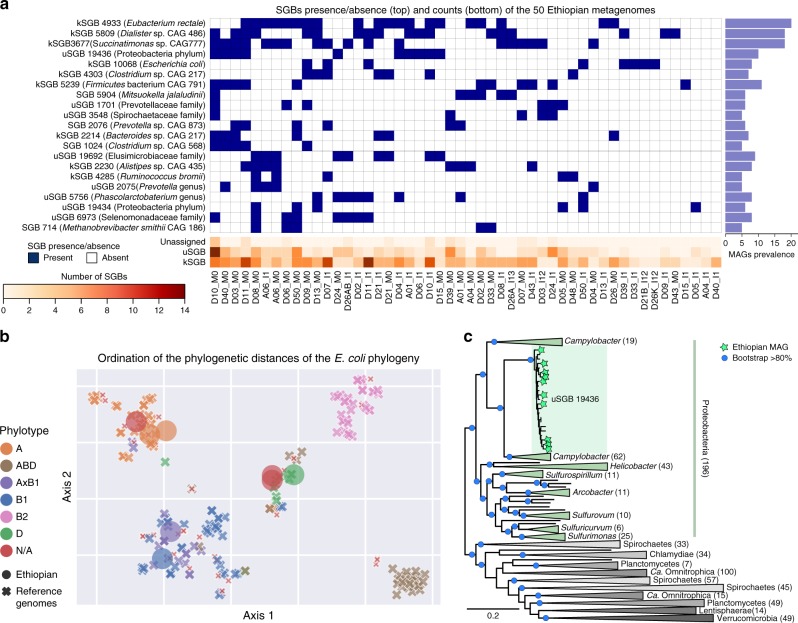
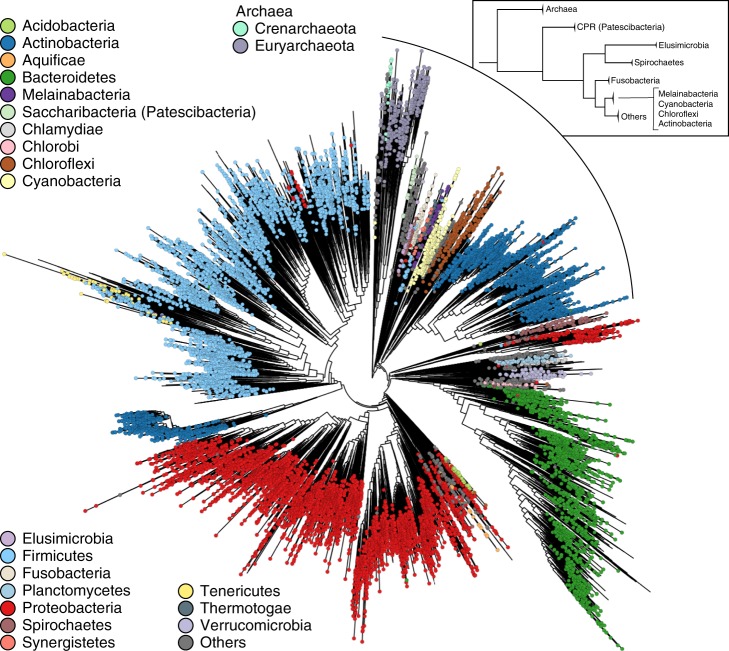
Microbial genomes are available at an ever-increasing pace, as cultivation and sequencing become cheaper and obtaining metagenome-assembled genomes (MAGs) becomes more effective. Phylogenetic placement methods to contextualize hundreds of thousands of genomes must thus be efficiently scalable and sensitive from closely related strains to divergent phyla. We present PhyloPhlAn 3.0, an accurate, rapid, and easy-to-use method for large-scale microbial genome characterization and phylogenetic analysis at multiple levels of resolution. PhyloPhlAn 3.0 can assign genomes from isolate sequencing or MAGs to species-level genome bins built from >230,000 publically available sequences. For individual clades of interest, it reconstructs strain-level phylogenies from among the closest species using clade-specific maximally informative markers. At the other extreme of resolution, it scales to large phylogenies comprising >17,000 microbial species. Examples including Staphylococcus aureus isolates, gut metagenomes, and meta-analyses demonstrate the ability of PhyloPhlAn 3.0 to support genomic and metagenomic analyses.
Conflict of interest statement
The authors declare no competing interests.
{kind=link}
{kind=link}
{kind=link}
{kind=link}