Stacks: building and genotyping Loci de novo from short-read sequences
- PMID: 22384329
- PMCID: PMC3276136
- DOI: 10.1534/g3.111.000240
Stacks: building and genotyping Loci de novo from short-read sequences
Abstract
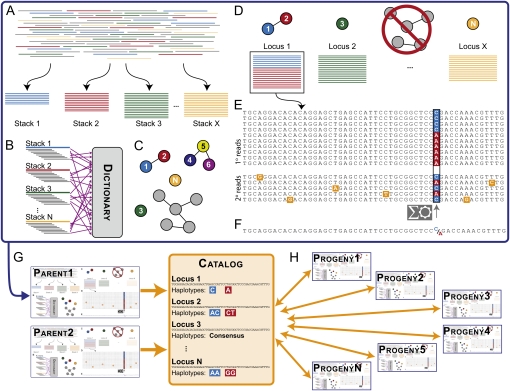
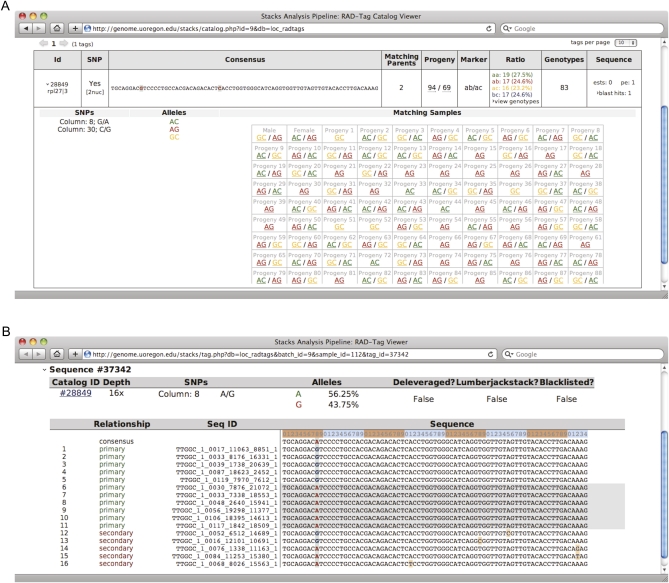
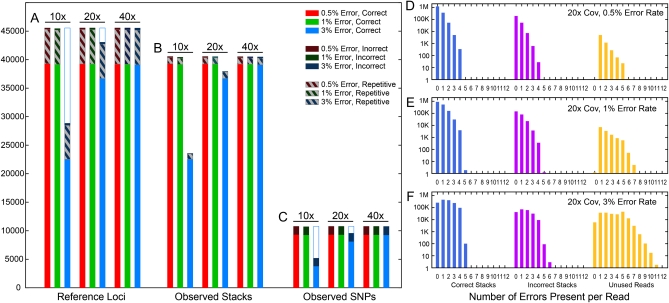
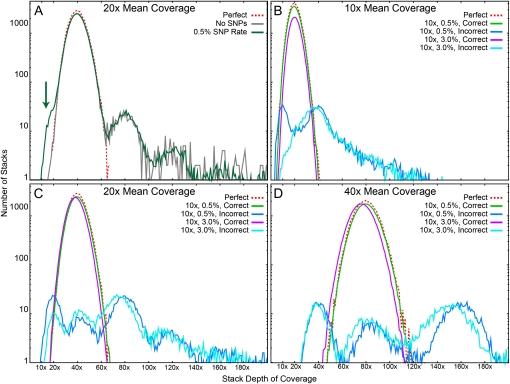
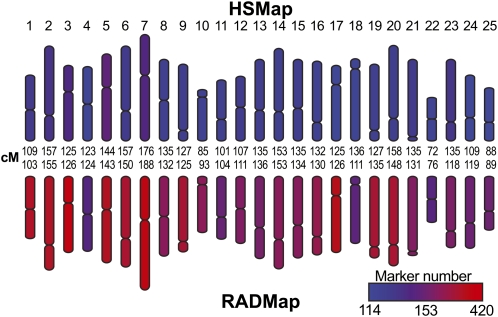
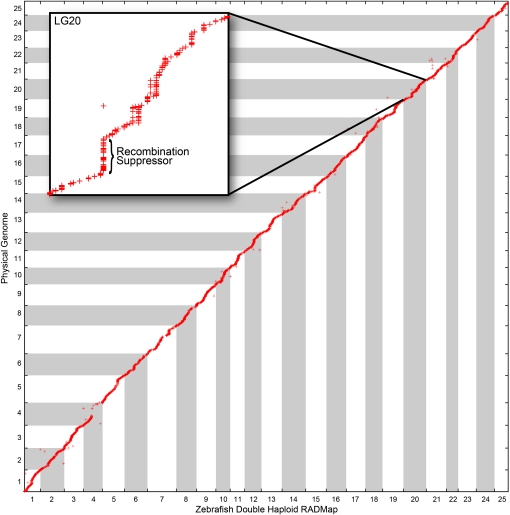
Advances in sequencing technology provide special opportunities for genotyping individuals with speed and thrift, but the lack of software to automate the calling of tens of thousands of genotypes over hundreds of individuals has hindered progress. Stacks is a software system that uses short-read sequence data to identify and genotype loci in a set of individuals either de novo or by comparison to a reference genome. From reduced representation Illumina sequence data, such as RAD-tags, Stacks can recover thousands of single nucleotide polymorphism (SNP) markers useful for the genetic analysis of crosses or populations. Stacks can generate markers for ultra-dense genetic linkage maps, facilitate the examination of population phylogeography, and help in reference genome assembly. We report here the algorithms implemented in Stacks and demonstrate their efficacy by constructing loci from simulated RAD-tags taken from the stickleback reference genome and by recapitulating and improving a genetic map of the zebrafish, Danio rerio.
Keywords: Illumina; RAD-seq; RAD-tag; meiotic linkage map; zebrafish.
Figures
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
References
-
- Amores A., Force A., Yan Y. L., Joly L., Amemiya C., et al. , 1998. Zebrafish hox clusters and vertebrate genome evolution. Science 282: 1711–1714 - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources