#Parser
Parser
#Parser
Parser
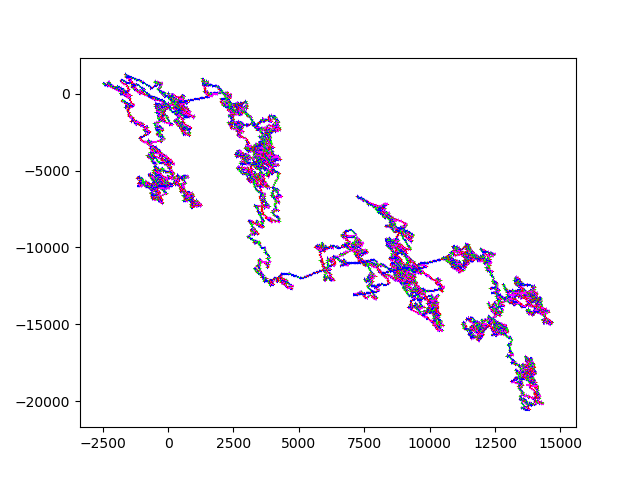
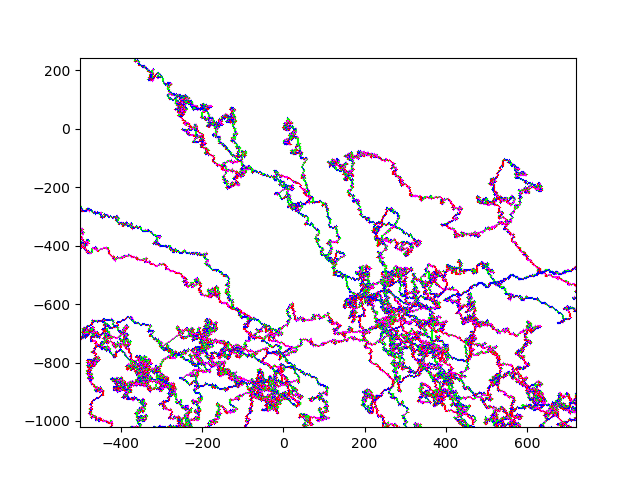
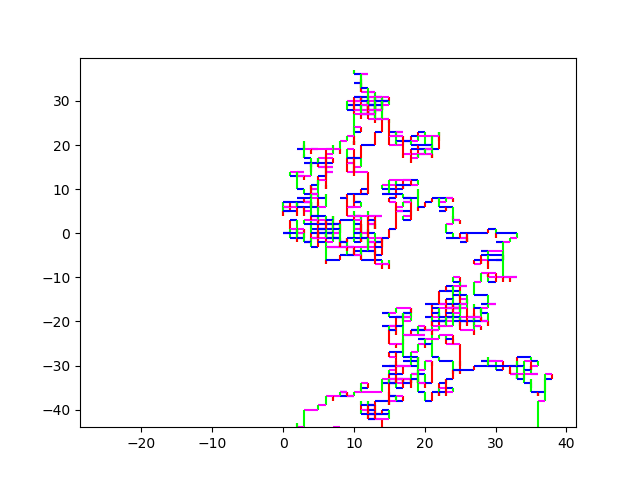
10M Strawberry.fna A zoom in on (0,0): Strawberry zoom And an even more useful / useless zoom onto (0,0): Even more Zoom
{kind=link}
{kind=link}
{kind=link}
10M Strawberry.fna A zoom in on (0,0): Strawberry zoom
10M Strawberry.fna A zoom in on (0,0): Strawberry zoom And an even more useful / useless zoom onto (0,0): Even more Zoom
#Parser
Your parser has a bug in line 62:
raw = ''.join([n for n in file.readlines() if not n.startswith('>')]).replace('\n', "").lower()
will concatenate all sequences, despite them potentially belonging to different DNA Sequences. Here is Wikipedia (the best source to cite xD):
A multiple sequence FASTA format would be obtained by concatenating several single sequence FASTA files. This does not imply a contradiction [...] hence forcing all subsequent sequences to start with a ">" [...].
You also don't respect the header (;) either. Further
Sequences may be protein sequences or nucleic acid sequences
Now my biology is lacking at best. I assume you are not respecting that because you can make the assumption to only ever deal with nucleic acids in DNA? Hence you ignore checking for that?
You seem to further seem to be able to reduce this to only A C G N T which I guess also has some very sound biological reason that I don't know.
This will probably blow up if you aren't careful what files you put in.
Large Array
There is multiple optimizations you could do, but only 1 that really makes sense here: Bin it. You don't need it.
You pre-compute a lot of information that you reuse exactly once and that you can compute on-the-fly. There is no benefit in having this. In fact, you are making it worse by reading the .fna once and then reading the array version of it, which is 40+ times bigger. Not cool :D
The short version of it is that largearray is wrapping python list()s which store 5-tupels (x,y,r,g,b) worth 40 byte per entry. strawberry.fna takes ~8.5 GB to represent in this format, whereas you can instead stick to a single character worth 1 byte and look-up the (r,g,b) value as well as compute (x,y) using the predecesor (which you know). This will save 40(!) times the disk/memory space (down to200 MB after optimization).
Here is a version that uses largearray, but saves only a character (worth 1 byte) instead of the tupel (40 byte): https://gist.github.com/FirefoxMetzger/9cfdf91f9b838f755d8ae68ab2a52ac8
I've also added numpy as dependency and refactored your code (a lot) because I couldn't make heads and tails of it.
Here is an even shorter version that no longer needs largearray:
https://gist.github.com/FirefoxMetzger/95b4f30fee91a21ab376cd4db9ccce42
I also removed the timing comments, because that can be done more efficiently with cProfile or another profiler of your choice. In this scenario, I felt like the prints obstructed the code a lot; now it's easier to read. If a blinking cursor with no output irritates you, feel free to add more prints (at the cost of readability and a tiny bit of runtime).
Output
I've ran the strawberry.fna and it tried to create a 94980x100286 image. That's ~9.5 Gpx (Gigapixel)! 29.5 GB as an uncompressed image is no fun. At least on my machine it doesn't fit into memory uncompressed. Similarly, if I look at your strawberry.png it doesn't seem to open correctly; I only get a reddish shadow.
On your machine (assuming you have a bigger one, since you can apparently fit 30+GB into memory without issues) you might get more out of this.
Fortunately most of these pixels are empty / transparent. In fact for strawberry.fna only 2.2% of the pixels are not transparent. Potentially even less, because I've noticed that they sometimes overlap, e.g. a "GC" sequence would only be visible as "C" sticking out to the side.
My attempt would be to use matplotlib, however that is still fairly slow and known to not cope too well with big data. It runs slightly faster then your initial version and the memory requirements are better; still its not enough for strawberries.fna on my machine. Yet, you get an interactive figure that you can zoom around in and "take screenshots". I like this way to explore the sequence better.
Another advantage is that you can specify file format and viewport, legends, axis and so forth.
Here is the code for that:
#!/usr/bin/env python3
# Python3
import logging
from argparse import ArgumentParser
from os import remove
from os.path import exists
from re import sub
from time import time
import numpy as np
import matplotlib.pyplot as plt
from matplotlib.collections import LineCollection
uuid = id(time())
parser = ArgumentParser()
parser.add_argument("file", help="Location of input file. path/to/file (FASTA file formats supported)")
parser.add_argument("-d", "--debug-file", help="Where to store debug file. path/to/file (Default ./cache/debug.log")
args = parser.parse_args()
filepath = args.file
if not exists(filepath):
raise Exception("Path of input file does not exist")
debugpath = '.cache/debug%d.log' % uuid
if args.debug_file:
debugpath = args.debug_file
if exists(debugpath):
remove(debugpath)
logging.basicConfig(filename=debugpath, level=logging.DEBUG)
logging.info("Init: %d" % uuid)
with open(filepath, 'r') as file:
logging.info("Serializing %s ..." % filepath)
raw = ''.join([n for n in file.readlines() if not n.startswith('>')]).replace('\n', "").lower()
raw = sub(r'[rykmswbdhv-]', "n", raw) # Handles miscellaneous FASTA characters
raw = sub(r'[^atgcn]', "", raw) # Remove everything except 4 bases and not-known
test_length = min(5e6, len(raw))
print("Processing %d elements. Have fun waiting! I'll go grab a coffee"
% test_length)
action_lookup = {
"a": np.array((0, -1)),
"t": np.array((0, 1)),
"g": np.array((-1, 0)),
"c": np.array((1, 0)),
"n": np.array((0, 0))
}
color_lookup = {
"a": (0, 1, 0),
"t": (1, 0, 0),
"g": (1, 0, 1),
"c": (0, 0, 1),
"n": (0, 0, 0)
}
fig, ax = plt.subplots()
lines = np.zeros((int(test_length), 2, 2), dtype=np.int16)
colors = list()
cursor = np.array((0, 0))
old_cursor = cursor.copy()
for idx, base in enumerate(raw):
if idx >= test_length:
break
# get the actions associated from dict
cursor += action_lookup[base]
lines[idx, 0, :] = old_cursor
lines[idx, 1, :] = cursor
colors.append(color_lookup[base])
old_cursor = cursor.copy()
print("Done gathering data - plotting now")
linesegments = LineCollection(
lines,
colors=colors
)
ax.add_collection(linesegments)
plt.plot()
plt.show()
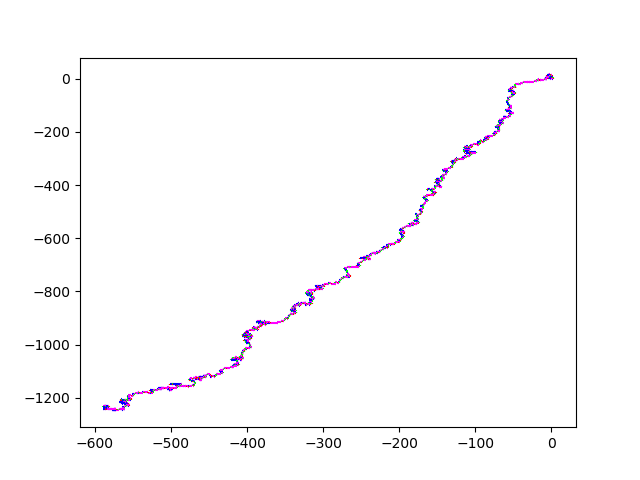
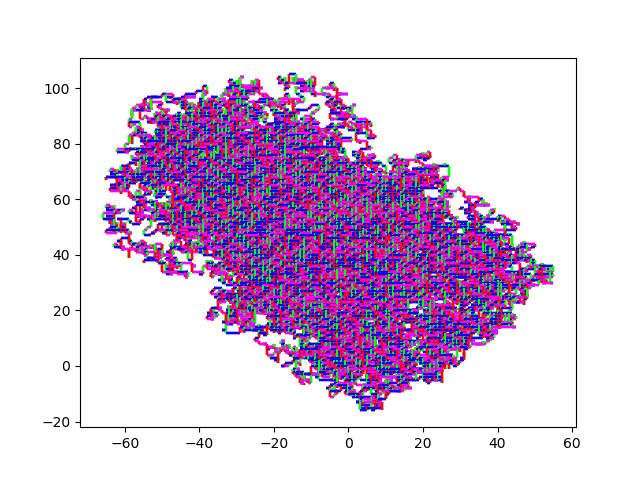
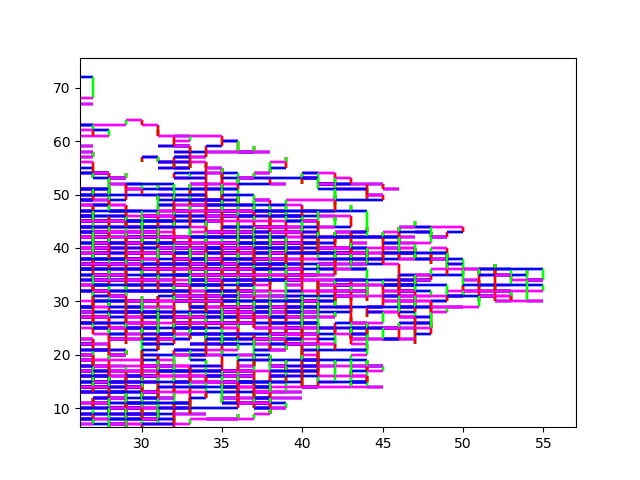
And here some sexy images. (They always start at (0,0)). HIV.fna: hiv.fna random2.fna: random2.fna Demonstrating the crop/zoom-ability on random2: Crop of Random2.fna
{kind=link}
{kind=link}
{kind=link}
I can also limit the amount of elements processed. That way I can visualize the first 10 million elements of strawberry.fna. I find this visualization to be of limited use, but I guess it makes more sense to you?
10M Strawberry.fna A zoom in on (0,0): Strawberry zoom