Predicting multiple conformations of ligand binding sites in proteins suggests that AlphaFold2 may remember too much
- PMID: 39565312
- PMCID: PMC11621821
- DOI: 10.1073/pnas.2412719121
Predicting multiple conformations of ligand binding sites in proteins suggests that AlphaFold2 may remember too much
Abstract
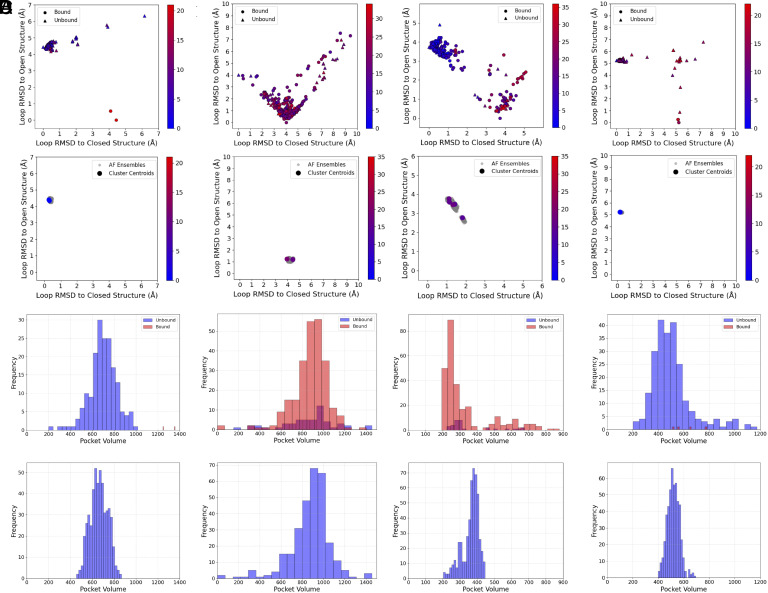
The goal of this paper is predicting the conformational distributions of ligand binding sites using the AlphaFold2 (AF2) protein structure prediction program with stochastic subsampling of the multiple sequence alignment (MSA). We explored the opening of cryptic ligand binding sites in 16 proteins, where the closed and open conformations define the expected extreme points of the conformational variation. Due to the many structures of these proteins in the Protein Data Bank (PDB), we were able to study whether the distribution of X-ray structures affects the distribution of AF2 models. We have found that AF2 generates both a cluster of open and a cluster of closed models for proteins that have comparable numbers of open and closed structures in the PDB and not too many other conformations. This was observed even with default MSA parameters, thus without further subsampling. In contrast, with the exception of a single protein, AF2 did not yield multiple clusters of conformations for proteins that had imbalanced numbers of open and closed structures in the PDB, or had substantial numbers of other structures. Subsampling improved the results only for a single protein, but very shallow MSA led to incorrect structures. The ability of generating both open and closed conformations for six out of the 16 proteins agrees with the success rates of similar studies reported in the literature. However, we showed that this partial success is due to AF2 "remembering" the conformational distributions in the PDB and that the approach fails to predict rarely seen conformations.
Keywords: binding hot spot; conformational change; machine learning; protein mapping; protein structure prediction.
Conflict of interest statement
Competing interests statement:The authors declare no competing interest.
Figures
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
References
-
- Nayal M., Honig B., On the nature of cavities on protein surfaces: Application to the identification of drug-binding sites. Proteins 63, 892–906 (2006). - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources