A comparison of marker gene selection methods for single-cell RNA sequencing data
- PMID: 38409056
- PMCID: PMC10895860
- DOI: 10.1186/s13059-024-03183-0
A comparison of marker gene selection methods for single-cell RNA sequencing data
Abstract
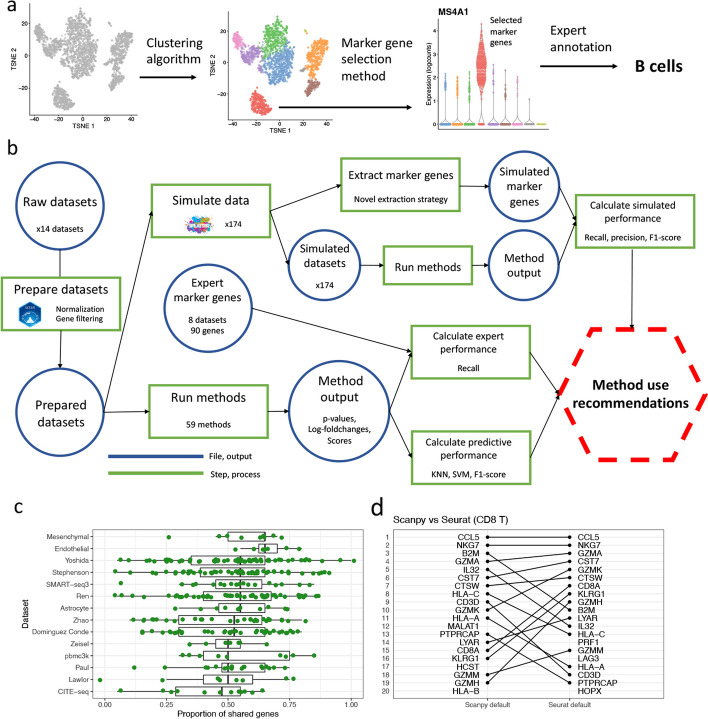
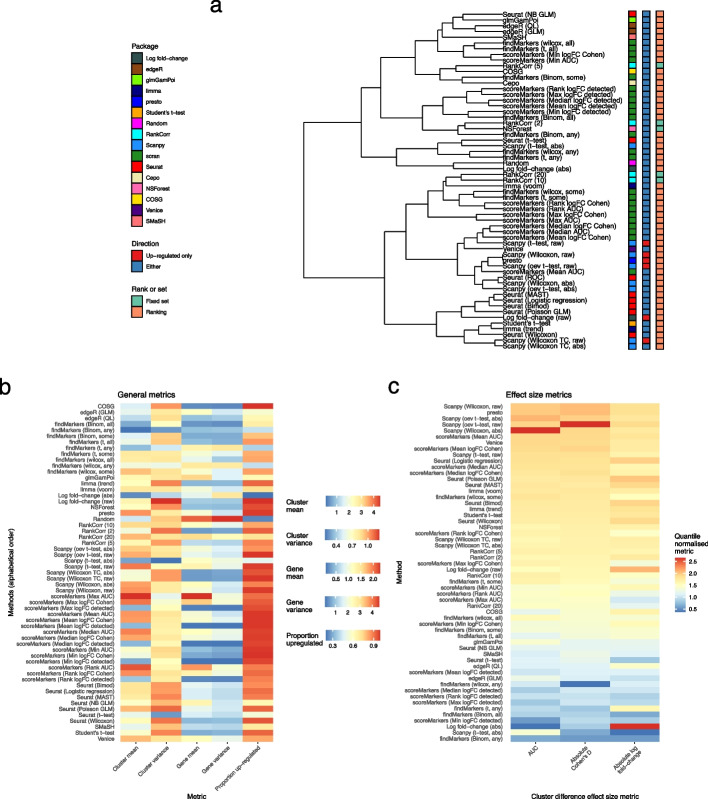
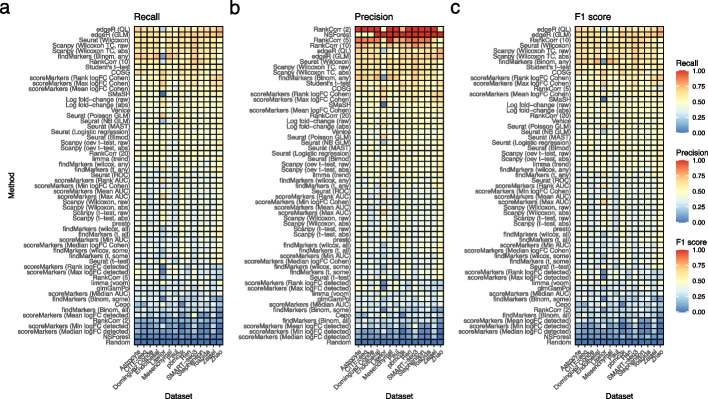
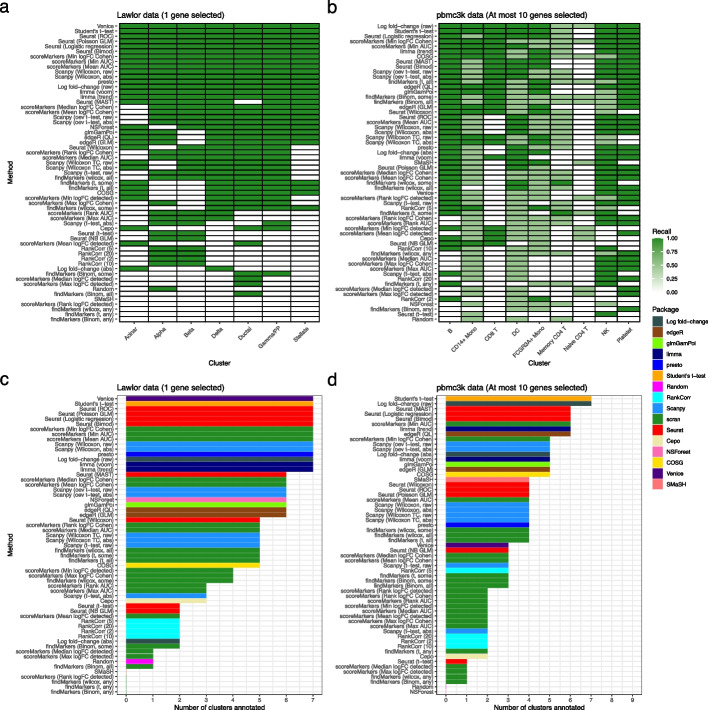
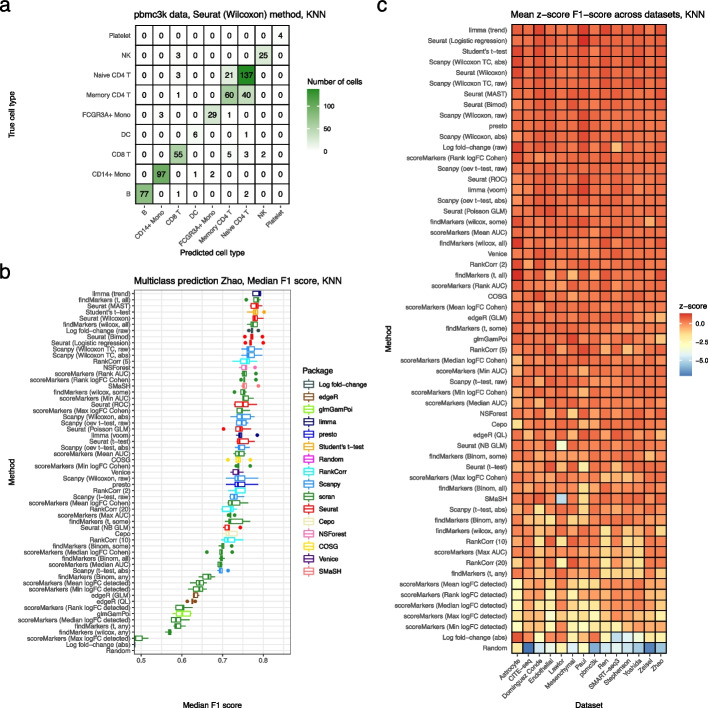
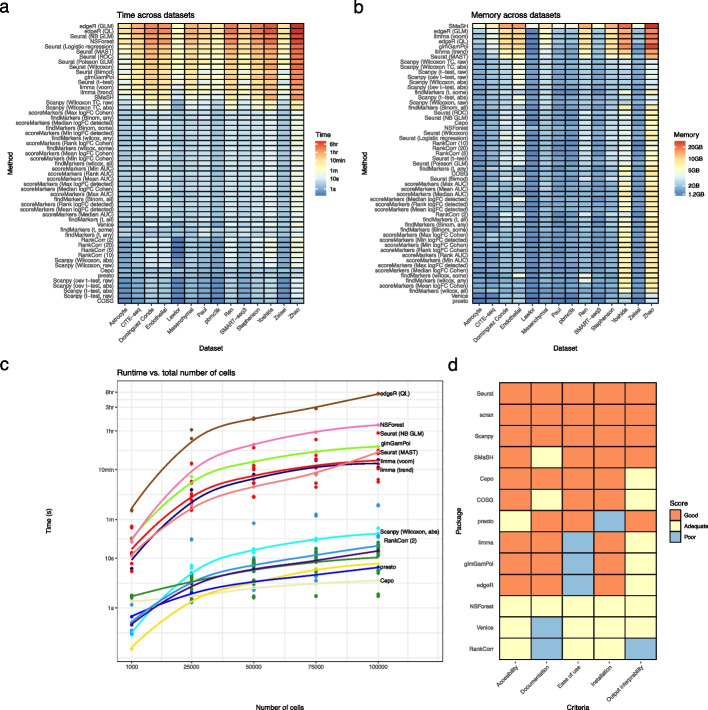
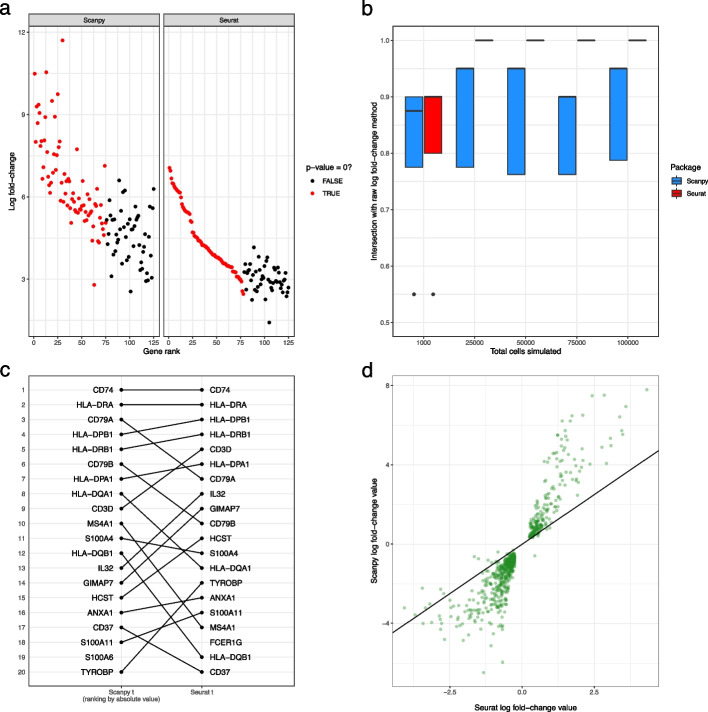
Background: The development of single-cell RNA sequencing (scRNA-seq) has enabled scientists to catalog and probe the transcriptional heterogeneity of individual cells in unprecedented detail. A common step in the analysis of scRNA-seq data is the selection of so-called marker genes, most commonly to enable annotation of the biological cell types present in the sample. In this paper, we benchmark 59 computational methods for selecting marker genes in scRNA-seq data.
Results: We compare the performance of the methods using 14 real scRNA-seq datasets and over 170 additional simulated datasets. Methods are compared on their ability to recover simulated and expert-annotated marker genes, the predictive performance and characteristics of the gene sets they select, their memory usage and speed, and their implementation quality. In addition, various case studies are used to scrutinize the most commonly used methods, highlighting issues and inconsistencies.
Conclusions: Overall, we present a comprehensive evaluation of methods for selecting marker genes in scRNA-seq data. Our results highlight the efficacy of simple methods, especially the Wilcoxon rank-sum test, Student's t-test, and logistic regression.
Keywords: Benchmarking; Bioinformatics; Single-cell.
© 2024. The Author(s).
Conflict of interest statement
While this manuscript was under consideration for publication DJM became an Editorial Board member of Genome Biology. JMP declares no competing interests.
Figures
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources