Spatially informed clustering, integration, and deconvolution of spatial transcriptomics with GraphST
- PMID: 36859400
- PMCID: PMC9977836
- DOI: 10.1038/s41467-023-36796-3
Spatially informed clustering, integration, and deconvolution of spatial transcriptomics with GraphST
Abstract
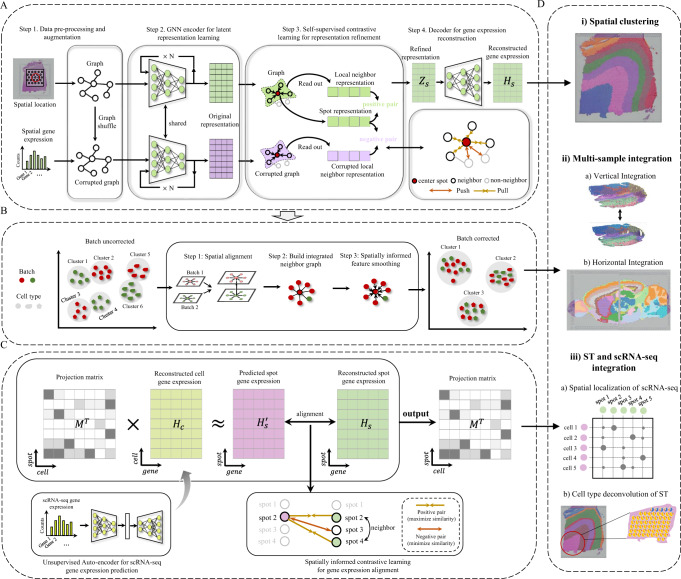
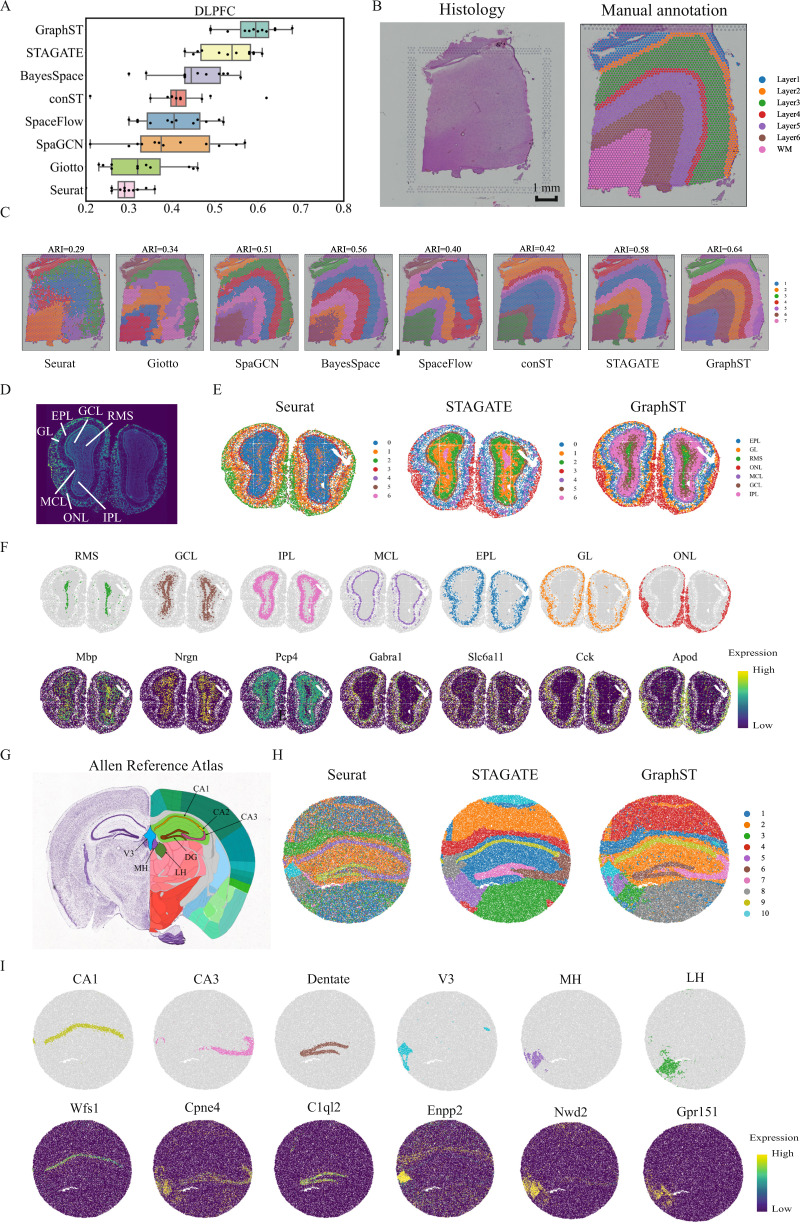
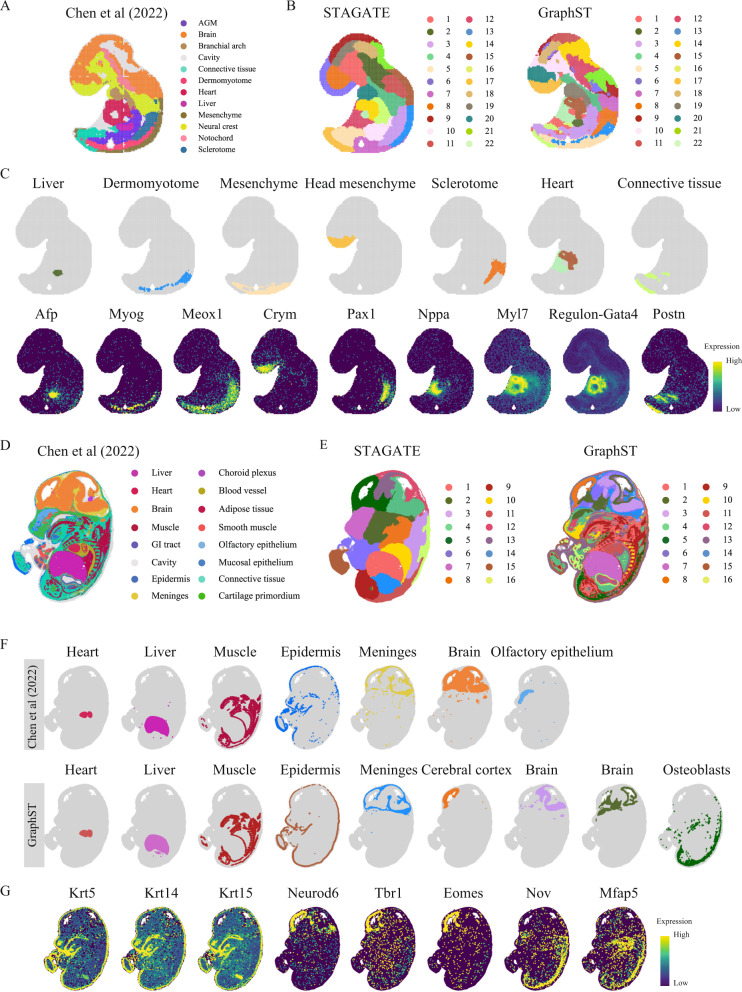
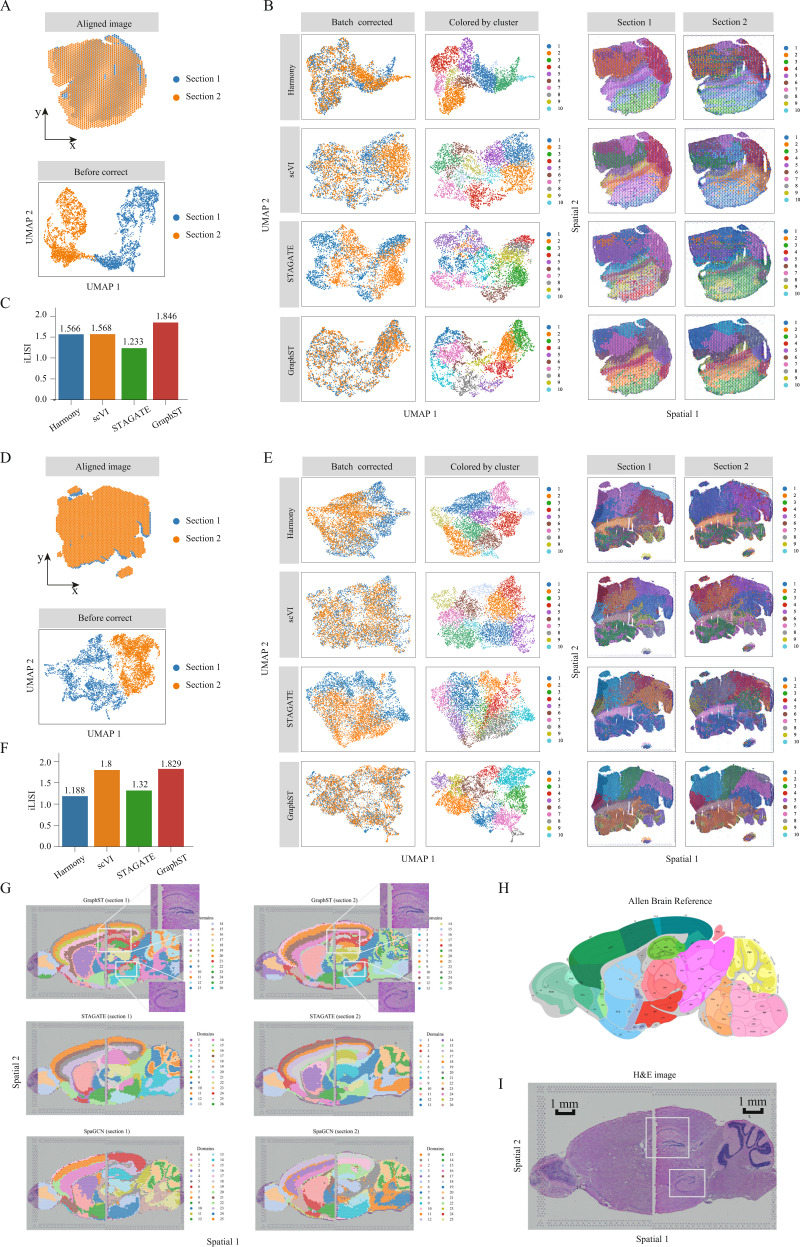
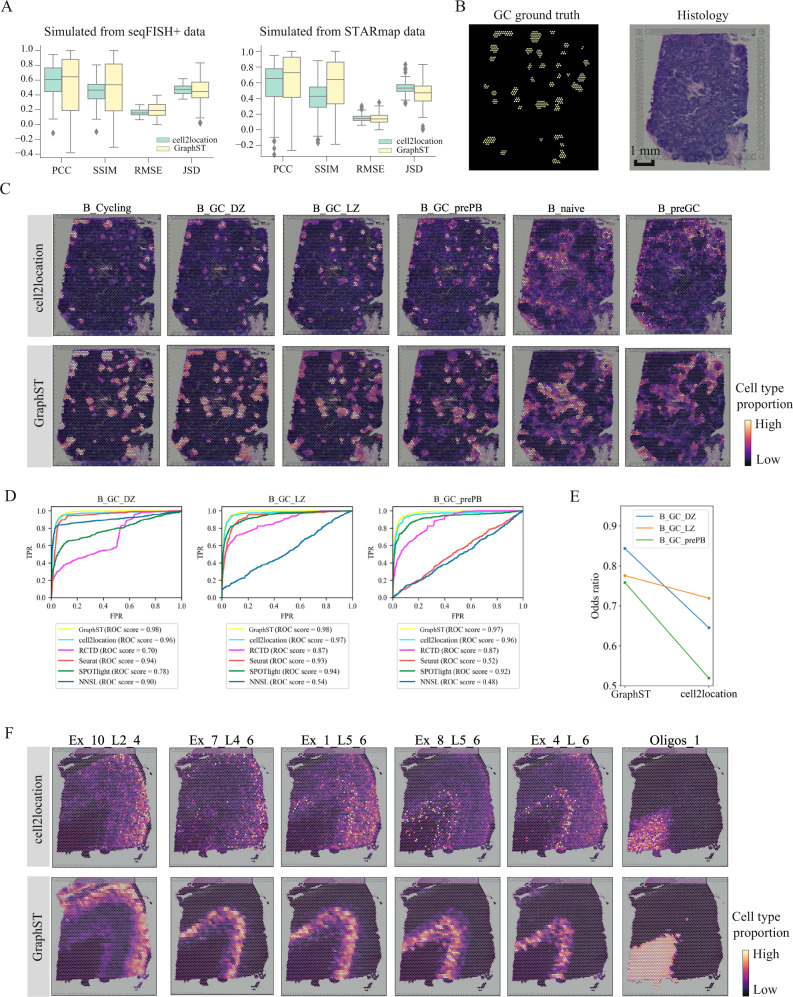
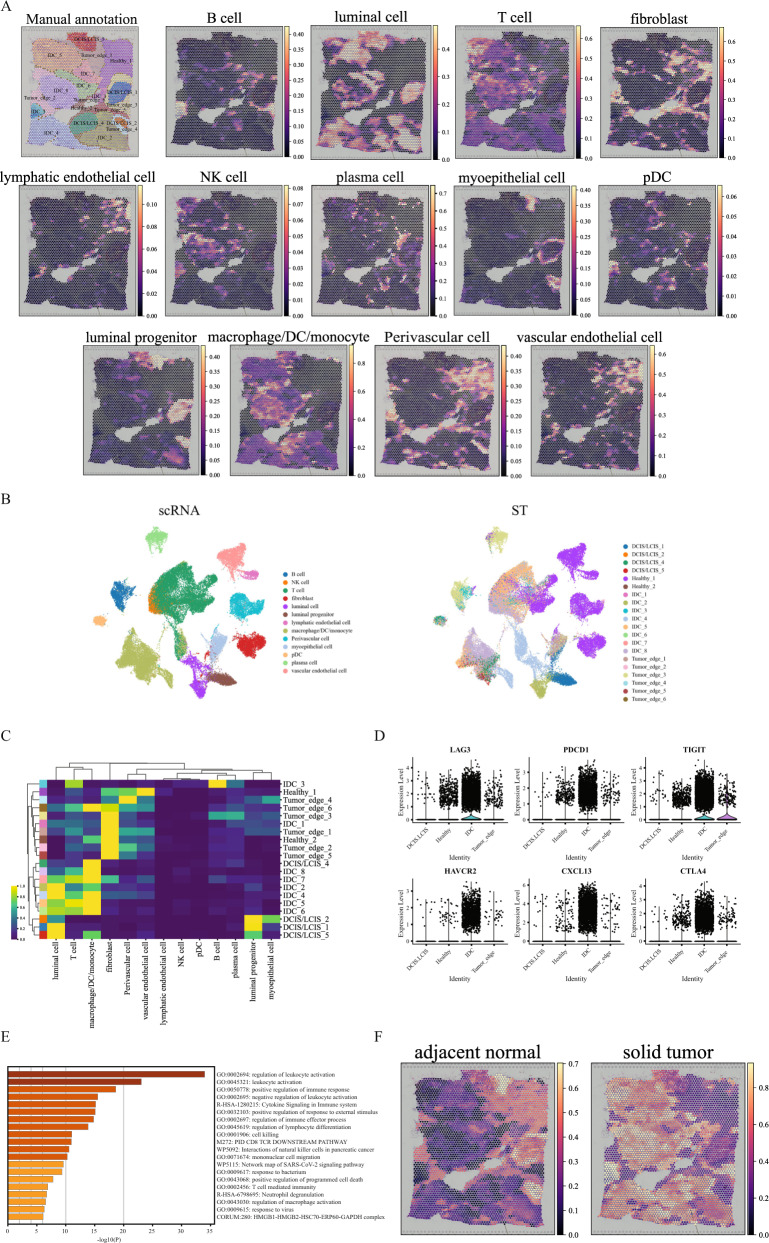
Spatial transcriptomics technologies generate gene expression profiles with spatial context, requiring spatially informed analysis tools for three key tasks, spatial clustering, multisample integration, and cell-type deconvolution. We present GraphST, a graph self-supervised contrastive learning method that fully exploits spatial transcriptomics data to outperform existing methods. It combines graph neural networks with self-supervised contrastive learning to learn informative and discriminative spot representations by minimizing the embedding distance between spatially adjacent spots and vice versa. We demonstrated GraphST on multiple tissue types and technology platforms. GraphST achieved 10% higher clustering accuracy and better delineated fine-grained tissue structures in brain and embryo tissues. GraphST is also the only method that can jointly analyze multiple tissue slices in vertical or horizontal integration while correcting batch effects. Lastly, GraphST demonstrated superior cell-type deconvolution to capture spatial niches like lymph node germinal centers and exhausted tumor infiltrating T cells in breast tumor tissue.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources