The latest release (version 2025.1) of the IUPHAR/BPS Guide to Pharmacology database has been made. Released on 2nd April 2025 this is the first release of the year. The following blog post gives details of the key content updates and website changes. GtoPdb now contains:
In this release, 181 new ligands, 178 new curated interactions have been added.
We have added 8 protein targets since release 2024.3, summarised in the table below:
Ligands
In total, 180 new ligands have been added to the GtoPdb since the last update in December 2024 (release 2024.4). These include synthetic small molecules, antibacterials, antibodies, peptides and natural products.
Our list of approved drugs for 2024 was expanded to a total of 61. This set includes first approvals from regulatory agencies other than the FDA and EMA, such as China’s NPMA and Japan’s PMDA. 7 of the new approvals are not included as fully-curated ligand entries as they fall out with our inclusion criteria. Presentation of this set of drugs on the website is discussed in more detail below, in the database development section.
The new WHO INN proposed list 132 (PL132) was published in mid-February 2025.
Of the 249 INNs, 54 are curated in the GtoPdb; 24 existed under alternate names and were matched on chemical structures to the INNS. A further 30 (the majority of which are kinase inhibitors) are brand new entities for the GtoPdb, and where possible the structures are matched to internal company IDs (even speculatively when no name-structure has been formally disclosed), patent claims, target-specific quantitative data and clinical trials. The INN secutrelvir was matched to the Shionogi SARS-CoV-2 Mpro inhibitor S892216.
GtoPdb ligands that have INNS from PL132 are listed in tables 1 and 2 below.
Table 1: INNs from PL132 for kinase inhibitors that are in the GtoPdb
Table 2: INNs from PL132 in the GtoPdb for miscellaneous protein targets
Our collaboration with Antibiotic DB (ADB; www.antibioticdb.com) continues to allow us to extend the coverage of ligands with annotated antibacterial activity in GtoPdb and provide comprehensive chemistry and pharmacology for select antibacterials curated within ADB, via reciprocal links. This project is supported by the Global Antibiotic Research and Development Partnership (GARDP; https://gardp.org/).
Currently we have 648 ligands tagged in GtoPdb as ‘antibacterial’ and 628 of these have links to compounds at ADB. 267 are approved drugs. Since our last release we have added 34 new antibacterial ligands including:
We are developing a new landing page for antibacterials in the Guide to Pharmacology and this will be publicly available as part of a future database release.
We have developed a new page that records details of drug approvals. https://www.guidetopharmacology.org/GRAC/DrugApprovalsForward
Each year, the Guide to Pharmacology Curation Team puts together a list of the latest approved drugs from FDA, EMA and MHRA as well as first-time approvals from other agencies. The new drug approval page, shows these approved drugs in table, organised by year of approval. The majority of these drugs will be curated in the database, and if so they are hyperlinked to their respective summary pages from the INN in the table. There are some cases were we have listed an approved drug but it doesn’t have a link because it will not have been curated.
The latest release (version 2024.4) of the IUPHAR/BPS Guide to Pharmacology database has been made. Released on 4th December 2024 this is the fourth release of the year. The following blog post gives details of the key content updates and website changes. GtoPdb now contains:
In this release, 60 new curated interactions have been added.
So far in 2024 826 new curated interactions have been added to the database, along with updates to 341 existing interactions.
We have added 3 protein targets since release 2024.3. These are supported with evidence of pharmacological tool modulators, and were added in conjunction with our natural products curation project.
Approved drugs
Since the 2024.3 release the FDA have approved 9 more drugs, bringing the total for the year so far to 49.
Use the Ligand ID link to view the ligand at the Guide to Pharmacology
Continuing to follow developments towards pan-coronavirus antivirals we have added new inhibitors SS148 and CoV nsp16 inhibitor 2a [PMID: 34257831].
These compounds inhibit both of the CoV methyltransferases (nsp16 and nsp14) that are essential for viral RNA capping and enabling the virus to evade immune detection in humans. SS148 has demonstrated broad-spectrum activity across human coronaviruses in vitro.
We now have all 18 of the currently approved drugs in this class in the GtoPdb. Unfortunately, for many of these we are unable to provide sull chemical structures, and are restricted to the available nucleic acid sequence information. We have added new fields to present both the sequences and targets for each of these drugs. In line with all of our other approved drugs we curate approval information (dates, indications, trade names etc). We are continuing to add drugs from this class that are clinical candidates in phase 1-3 development.
Our collaboration with Antibiotic DB (ADB; www.antibioticdb.com) continues to allow us to extend the coverage of ligands with annotated antibacterial activity in GtoPdb and provide comprehensive chemistry and pharmacology for select antibacterials curated within ADB, via reciprocal links. This project is supported by the Global Antibiotic Research and Development Partnership (GARDP; https://gardp.org/).
Currently we have 613 ligands tagged in GtoPdb as ‘antibacterial’ and 589 of these have links to compounds at ADB. 260 are approved drugs. Since our last release we have added 18 new antibacterial ligands including:
A reminder of some new pages and updates made at our last release (2024.3):
Our latest database report provides an overview of progress and status of the IUPHAR/BPS Guide to PHARMACOLOGY (GtoPdb) for the period April 2024 to October 2024.
Click to access GtoPdb_Database_Report_Nov_2024.pdf
It can also be access at our Zenodo repository https://doi.org/10.5281/zenodo.14046004
The latest release (version 2024.3) of the IUPHAR/BPS Guide to Pharmacology database has been made. Released on 3rd October 2024 this is the third release of the year. The following blog post gives details of the key content updates and website changes. GtoPdb now contains:
In this release, 119 new curated interactions have been added.
So far in 2024 761 new curated interactions have been added to the database, along with updated to 338 existing interactions.
3 new protein targets have been added to the Guide since our last update.
A range of new ligands have been curated from analysis of INN proposed list 131 (August 2024)
In total 28 synthetic small molecules with INNs in this list are now included in the GtoPdb; 6 matched chemical structures that were already in our data set (the top 6 in the table below), and 22 are brand new entries (LIDs >13480). Where possible the INNs have been linked to (probable) company research codes, corroborating publications, quantitative interaction data, and clinical trials. 18/28 are kinase inhibitors, 2 are EZH2 inhibitors, 1 is a PROTAC, and the others are intended to modulate a range of therapeutic targets including SLC6A19, cannabinoid, orexin and opioid receptors, COX-2 and autotaxin. Analysis is continuing to identify any of the monoclonal antibodies that are directed against proteins that are not already included in the GtoPdb, to expand our coverage of emerging drug targets.
Use the Ligand ID link to view the ligand at the Guide to Pharmacology
Approved drugs
With access to the EMA’s master EPAR download file restored (it had been unavailable since Dec 2023), we were able to retrospectively update the EU approval data for >30 drugs. In addition, the FDA has approved 16 new drugs since our last update, 14 of which meet the inclusion criteria for the GToPdb. We have also included 2 new drugs that were approved in jurisdictions outside of the US and EU.
Our collaboration with Antibiotic DB (ADB; www.antibioticdb.com) continues to allow us to extend the coverage of ligands with annotated antibacterial activity in GtoPdb and provide comprehensive chemistry and pharmacology for select antibacterials curated within ADB, via reciprocal links. This project is supported by the Global Antibiotic Research and Development Partnership (GARDP; https://gardp.org/).
Currently we have 594 ligands tagged in GtoPdb as ‘antibacterial’ and 566 of these have links to compounds at ADB. 255 are approved drugs.
Since our last release we have added 31 new antibacterial ligands including:
The latest release (version 2024.2) of the IUPHAR/BPS Guide to Pharmacology database has been made. Released on 26th June 2024 this is the second release of the year. The following blog post gives details of the key content updates and website changes. GtoPdb now contains:
The Adrenoceptors family has been widely updated. This includes a completely revised detailed introduction, and additions to ligands, interactions and other tables on all nine of the receptor subtypes pages. The updated material was provided by the Adrenoceptors subcommittee, Roger J. Summers, Martin C. Michel and Jillian G. Baker.
19 new protein targets have been added to the Guide since our last update. They represent molecular targets for cancer, inflammation, immunity, viral infection and Parkinson’s disease. Experimental tool compounds or clinical candidates that modulate protein functions have been curated where available.
Our collaboration with Antibiotic DB (ADB; www.antibioticdb.com) continues to allow us to extend the coverage of ligands with annotated antibacterial activity in GtoPdb and provide comprehensive chemistry and pharmacology for select antibacterials curated within ADB, via reciprocal links. This project is supported by the Global Antibiotic Research and Development Partnership (GARDP; https://gardp.org/).
Currently we have 562 ligands tagged in GtoPdb as ‘antibacterial’ and 537 of these have links to compounds at ADB. Since our last release we have added 25 new antibacterial ligands including:
A new release, version 2024.1, has been made of the IUPHAR/BPS Guide to Pharmacology database. Released on 26th March 2024 this is the first release of the year. The following blog post gives details of the key content updates and website changes. GtoPdb now contains:
11 new protein targets have been added to the GtoPdb since our last update. Ten of these are enzymes, with SIGLEC15 being the only new non-enzyme protein. PPP1CA, PPP2CA and ZDHHC3 were added as part of our natural products project. The remainder were added when new ligands for the targets were identified from literature searches
microcystin-LR, calyculin A)
Release of the newest proposed INN list (PL130) in February provided the opportunity to curate new nominally clinically-relevant entities. There were 414 INNs in this list. So far we have curated 27 kinase inhibitors from PL130, and where possible we have endeavoured to match the INNs to research codes in disclosed clinical pipelines, primary literature sources and clinical trials. We hope that this depth of curation provides additional insight and information for users of the GtoPdb. Occasionally the INNs match ligand structures that we have previously curated from other sources. Of note in PL130 was the inclusion of INNs for PROTAC type degrader molecules (zomiradomide, lirodegimod), and a kallikrein B1 gene editing agent (lonvoguran), both of which are relatively new classes of clinical interventions. We will continue to analyse this list to see if we can identify either new drug targets, or new pharmacological modalities for existing targets.
Drug approvals
Since the beginning of 2024 the FDA have approved 7 new drugs: as can be seen in the table below all but two of these are curated in the GtoPdb.
n/i (not included_ as the agent does not meet the current criteria for inclusion in the GtoPdb.
Our collaboration with Antibiotic DB (ADB; www.antibioticdb.com) continues to allow us to extend the coverage of ligands with annotated antibacterial activity in GtoPdb and provide comprehensive chemistry and pharmacology for select antibacterials curated within ADB, via reciprocal links. This project is supported by the Global Antibiotic Research and Development Partnership (GARDP; https://gardp.org/).
Currently we have 537 ligands tagged in GtoPdb as ‘antibacterial’ and 512 of these have links to compounds at ADB. Since our last release we have added 30 new antibacterial ligands including:
Published as a special issue in the British journal of Pharmacology every 2 years, The Concise Guide to PHARMACOLOGY provides brief overviews of the key properties of over 1,800 human drug targets and their pharmacology, and is a view of the data in the IUPHAR/BPS Guide to PHARMACOLOGY that allows side by side comparison of members of families of drug targets.
As a go to place for finding the best tools to use in the laboratory for any targets you are working on, and as an up to date list of the key reference material to read, there is no better resource for a pharmacologist.
The broad coverage includes 3 sections on ion channels, which will be of interest to physiologists. For many examples, molecular data are correlated with ion conductances at the cellular and channel level. Additionally, the large section on enzymes, with a major focus on kinases, peptidase and proteinases, will be relevant for biochemists and cell biologists wishing to identify whether selective tools for their enzyme of interest are available.
For those who teach pharmacology, the Concise Guide provides a handy starting point for researching specific pharmacological targets (receptors, ion channels, transporters, enzymes and other targets) which may be identified in lectures and practical classes. The further reading highlighted for these targets will allow greater depth of insight from authoritative sources.
Use it as a teaching resource and make sure your students have free access:
www.guidetopharmacology.org/concise
For those new to a research topic, the Concise Guide allows a ready comparison of individual pharmacological targets within a family.
Overview – Each family is introduced with an Overview, which includes the nomenclature status of the family and general data, for example on the endogenous ligands for the particular receptor family.
Table of Targets – Each table of targets includes clickable links to HUGO Gene Nomenclature Committee (HGNC) and UniProt, free online databases of the gene and protein information.
Where available, the table identifies the pharmacology (the most selective tools available) of that individual molecular target, with agonists, antagonists, substrates, products, and labelled ligands listed.
Comments sections underneath the tables identify generic or species variation and any association of targets with inherited disorders, together with any limitations of the pharmacological tools.
Further reading focusses on published reviews of the target family, highlighting the most relevant and recent publications, allowing the reader to identify further relevant resources for increasing the depth and breadth of their understanding.
This post has been copied and re-produced from the British Journal of Pharmacology website (https://bpspubs.onlinelibrary.wiley.com/journal/14765381/concise_guide_to_pharmacology.html) in order to publicise the Concise Guide to Pharmacology directly to followers of this blog and users of the IUPHAR/BPS Guide to Pharmacology.
The IUPHAR/BPS Guide to Pharmacology is delighted to be named as one of 15 new Global Core Biodata Resources (GCBRs) by the Global Biodata Coalition (GBC).
The GBC announced the outcome of its 2023 selection round for GCBRs on 11th December 2023 – you can read about the announcement here.
GtoPdb is proud to meet the stringent eligibility criteria that were part of the selection process. This covered scientific focus, size and reach of the user communities, quality of service, governance and impact on global research.
The inclusion of the 15 new resources brings the total number of GCBrs to 52. The full list of GCBRs can be viewed here.
The GBC defines biodata resources as any biological, life science, or biomedical database that archives research data generated by scientists, or functions as a knowledgebase by adding value to scientific data by aggregation, processing, and expert curation. Global Core Biodata Resources are biodata resources that are of fundamental importance to the wider biological and life sciences community and the long term preservation of biological data. They:
- provide free and open access to their data,
- are used extensively both in terms of the number and distribution of their users,
- are mature and comprehensive,
- are are considered authoritative in their field,
- are of high scientific quality, and
- provide a professional standard of service delivery.
Through its GCBR designation, the GBC seeks to draw the attention of life science funders to the most critical of the global set of biodata resources and to better understand the challenges and needs for biodata resource long-term sustainability. The GCBRs are deposition databases and knowledgebases of fundamental importance to the global life sciences and biomedical research communities, providing open access and long-term preservation of key biological data. Sustained, long-term support for these databases is paramount, as their discontinuation would have a highly detrimental impact on the global research endeavour.
The latest release of the IUPHAR/BPS Guide to Pharmacology database was made on 29th November 2023. This database release is version 2023.3, and is the third release this year. The following blog post gives details of the key content updates and website changes. GtoPdb now contains:
In October 2023 our latest NAR Database Issue update was published:
Harding SD, Armstrong JF, Faccenda E, Southan C, Alexander SPH, Davenport AP, Spedding M, Davies JA. (2023) The IUPHAR/BPS Guide to PHARMACOLOGY in 2024. Nucl. Acids Res. 2023 Oct 28:gkad944. Online ahead of print. doi: 10.1093/nar/gkad944. [Full text]. PMID: 37897341.
In this update we report on recent developments to the resource and describe expansion in content over the six database releases made during the last two years. The database update section of this paper focuses on two areas relating to important global health challenges. The first, SARS-CoV-2 COVID-19, remains a major concern and we describe our efforts to expand the database to include a new family of coronavirus proteins. The second area is antimicrobial resistance, for which we have extended our coverage of antibacterials in partnership with AntibioticDB, a collaboration that has continued through support from GARDP.
In November, our latest Database Report became available, covering in more detail the work we have undertaken in terms of curation and development, alongside usage statistics in the last six months GtoPdb_Database_Report_Nov_2023.pdf. Our report is accessible via our Zenodo repository: https://doi.org/10.5281/zenodo.10078018
New targets since Release Version 2023.2: We have curated a range of new targets and we highlight novel ligands that modulate their functions, and any therapeutic potential associated with pharmacological target manipulation.
Since our last update in August 2023, the FDA have approved 20 new drugs (58 in total so far for 2023, so ahead of the 52 for all of 2022). Only 3 of these did not meet the criteria to be included in GtoPdb
We have been keeping up to date with the progress of SARS-CoV-2 antiviral development by the COVID Moonshot open science project. In collaboration with the non-profit Drugs for Neglected Diseases initiative a clinical lead, DNDi-6510, has been selected, although its chemical structure has not yet been disclosed. We have however included a selection of Moonshot-discovered Mpro inhibitors as examples of their endeavours, and more details are available in the 2023 publication PMID: 37943932
The Moonshot Mpro inhibitors in GtoPdb are:
On the theme of SARS-CoV-2 antiviral development, we have included some new ligands that are reported as inhibitors of the CoV helicase (nsp13). Since the helicase is a crucial component of the CoV replication machinery, its inhibition is predicted to offer antiviral potential. Nsp13 is also reported to reduce the host interferon response, so limiting this effect would also likely contribute to COVID-19 therapeutic efficacy. To enhance nsp13’s inclusion we generated a new target entry (nsp13), to permit curation of quantitative ligand-target interaction data in the format used throughout other sections of the GtoPdb.
Nsp13 modulators curated are:
compound 4b [Ramsey et al., 2023]
Our collaboration with Antibiotic DB (ADB; www.antibioticdb.com) continues to allow us to extend the coverage of ligands with annotated antibacterial activity in GtoPdb and provide comprehensive chemistry and pharmacology for select antibacterials curated within ADB, via reciprocal links. This project is supported by the Global Antibiotic Research and Development Partnership (GARDP; https://gardp.org/).
Currently we have 507 ligands tagged in GtoPdb as ‘antibacterial’ and 488 of these have links to compounds at ADB. Since our last release we have added 35 new antibacterial ligands including:
The latest release of the IUPHAR/BPS Guide to Pharmacology database was made on 7th August 2023. This database release is version 2023.2, and is the second release this year. The following blog post gives details of the key content updates and website changes. GtoPdb now contains:
Since our last release in April we have added 14 new protein targets with clinical relevance, that have been identified from primary literature. They all have associated ligands which are also new additions. The majority of these new targets are being investigated for applications in different cancers as you can see in the table below.
A range of new ligands have been added. In addition to new information contributed by our target family subcommittees, additional sources for new ligands include primary med chem literature, patents, company pipeline analysis, INN lists, structure disclosures from scientific meetings and scanning of clinical trials for novel disease-associated agents.
Our PROTACs, molecular glues and other degraders ligand family (https://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=1030) has been expanded to over 90 experimental and investigational compounds, which in total target over 45 mainly human proteins, but we have two PROTACS that target the coronavirus 3CL-protease (Mpro).
Since our last update we have annotated 22 newly approved drugs – 15/22 are fully curated in the Guide. The other 7 don’t meet our selection criteria (e.g. target is non-human or is non-protein, imaging reagent, non-specific MMOA).
We are pleased to report that the Global Antibiotic Research and Development Partnership (GARDP; https://gardp.org/) has extended their funding of our collaboration with Antibiotic DB (ADB; www.antibioticdb.com) for a further 2 years. Through this interaction, GtoPdb provides chemistry and pharmacology for a curated set of antibacterial compounds with links to ADB.
Currently we have 471 ligands tagged in GtoPdb as ‘antibacterial’ and 455 of these have links to compounds at ADB. The antibacterials in the GtoPdb include approved drugs, WHO essential Medicines-listed medicines and a number of investigational and experimental compounds.
The latest release of the IUPHAR/BPS Guide to Pharmacology database was made on 26th April 2023. This database release is version 2023.1, and is the first release this year. The following blog post gives details of the key content updates and website changes. GtoPdb now contains:
Updates have been made to many target family summary pages in preparation for generating the 2023-24 issue of the Concise Guide to PHARMACOLOGY (due for publication at the end of the year). More significant modifications have been made to the summary pages for the GABAA receptors (in Ion channels) and Orexin receptors (in GPCRs). A major overhaul of the Taste 2 GPCRs (Bitter taste receptors) has been completed, with newly annotated details for ligands that interact with these receptors. We thank all of our contributors who have given so freely of their time to make these updates possible.
As a regular review of the INN lists from the WHO, we added 31 new kinase inhibitors to the Guide (see table 1), that were included in the Jan 2023 list of proposed INNs (List 128). The chemical structures allowed us to connect around half of these (16/31) INNs to clinical leads (some with declared name>structure), and others to patents from which we could obtain otherwise unpublished target information and interaction data. Five of the structures matched ligands that were already in the Guide. In total we have found interaction data for 27/31 of these inhibitors. For a few, we were unable to track down very little additional information.
A new set of antivirals for SARS-CoV-2 have been curated, based on patent extractions by our collaborator Chris Southan:
example I-1 [WO2023283831] (12588), TBK845 (12589), example 18 [WO2022013684] (12590), GDI-036 (12591), example 45 [WO2022229458] (12592) and compound 6 [WO2022133588] (12593) are Mpro inhibitors, and PLpro inhibitor 7 (12596) inhibits the other crucial coronavirus protease PLpro. New to this list of antivirals are two PROTAC class protein degraders, both of which target the Mpro for proteosomal degradation; these are CoV Mpro CRBN PROTAC 9 [US11530195] (21594) and CoV Mpro VHL PROTAC 9 [US11518759] (12595).
Keeping up with drug approvals for 2023, there have been 14 new drugs approved by the FDA since the beginning of the year. The table below provides the ligand IDs for the 11/14 that are curated in the Guide.
We are pleased to report that the Global Antibiotic Research and Development Partnership (GARDP; https://gardp.org/) has extended their funding of our collaboration with Antibiotic DB (ADB; www.antibioticdb.com) for a further 2 years. Through this interaction, GtoPdb provides chemistry and pharmacology for a curated set of antibacterial compounds with links to ADB.
Currently we have 425 ligands tagged in GtoPdb as ‘antibacterial’ and 413 of these have links to compounds at ADB. The antibacterials in the GtoPdb include approved drugs, WHO essential Medicines-listed medicines and a number of investigational and experimental compounds.
We have updated the version of ChEMBL used on the website. This is now at ChEMBL version 32. On the Guide to Pharmacology website we use data from ChEMBL to supplement both our pharmacology search and ligand activity charts.
We have updated our PubChem CID outlinks – updating ~380 links some of which had been to deprecated CIDs.
We have also done a check and update of our Ensembl out-links, updating deprecated ID/links in a number of cases.
We wish to offer our congratulations to Professor Jamie Davies, Professor of Experimental Anatomy, University of Edinburgh, who has been elected to the Cymdeithas Ddysgedig Cymru/Learned Society of Wales. The society’s new Fellows include academics from across Welsh and UK universities, writers, researchers and leaders from the world of higher education, as well as law, medicine and the media
Jamie has led the IUPHAR/BPS Guide to PHARMACOLOGY database since 2014, having taken over from Professor Tony Harmar, and has been instrumental in the launch of IUPHAR Guide to IMMUNOPHARMACOLOGY and IUPHAR/MMV Guide to Malaria PHARMACOLOGY. Professor Davies said, “I am delighted to be elected, and hope to play my small part in helping to promote the sciences in the land of my birth.”
Also elected to the society is Professor Clare Bryant, Professor of Innate Immunity, Cambridge University. Professor Bryant specialises in the field of innate immunity, which is one of the first lines of defence against antigens, harmful materials, entering the body. This has led to work developing medicines for treating allergies and Alzheimer’s disease. She was also instrumental in supporting the establishment of the IUPHAR Guide to IMMUNOPHARMACOLOGY.
Both Jamie and Clare are 2 of 5 new Fellows elected under the medicine and medical sciences speciality, and you can read more about them here: https://www.learnedsociety.wales/introducing-our-new-fellows-2023-stemm-1-category
Well done Jamie and Clare!
This post on SID tagging has been reproduced, with permission, from Dr. Chris Southan’s original post in his blog – Bio <-> Chem.
It is intended to be user-orientated, those interested in the technicalities are welcome to contact the Guide to Pharmacology curation team. The links and counts in this post we taken on 19th Dec 2022 and reflect data from the 2022.4 GtoPdb release.
Looking at the tags we have introduced will make this clearer. For starters, as many may know, as of release 2022.4 GtoPdb has 11603 PubChem substance (SID) records. Looking at SID472319339 we can find the following comments.
This includes the explicit tags
gtopdb_approved – Substance is an approved drug in GtoPdb.
gtopdb_antibacterial – Substance is tagged as an antibacterial in GtoPdb.
So we can employ an interface selection for the gtopdb_approved tag to get 1813 “approved” (you don’t usually need the [comment] as a field restrict but useful for more complex queries).
Similarly we can select as per below for the 365 antibacterial
We can then make the requisite intersect via the “Advanced” option on the query interface as below:
The intersection search is thus (“IUPHAR/BPS Guide to PHARMACOLOGY”[All Fields] AND gtopdb_antibacterial[comment]) AND (“IUPHAR/BPS Guide to PHARMACOLOGY”[All Fields] AND gtopdb_approved[comment]) with the result of 146
We have introduced an additional three selects. The first is
gtopdb_immuno – Substance is curated in IUPHAR Guide to Immunopharmacology (GtoImmuPdb)
with 1384 SIDs as per below:
Cutting the story short we can easily spot the difference between the small-molecule entries that will be subsumed into Compound (CIDs) and the “Structure not Available” entries (two above) that, as large macromolecules, cannot form CIDs and will thus remain SID-only entries
These are selectable via our fourth tag
gtopdb_antibody – GtoPdb identifies this substance as an Antibody
With which we can select the 335 antibody entries below :
As performed above the intersect below gives the 127 approved antibodies:
Last but not least our fifth tag is
gtopdb_malaria – Substance is curated in IUPHAR/MMV Guide to Malaria Pharmacology (GtoMPdb)
Combining with Venny
Either by combining queries or (much easier) using the query history under the advanced tag we can make any sequence of Boolean selects (i.e. AND, OR, NOT) from combinations of our five tags for “approved” “immuno” “antibiotic” “antibody” or “antimalarial”. Below we can see another way of slicing and dicing via Venny. The example result shown below compares three of the tag lists we have already looked at.
Another advantage of these GtoPdb specific retrievals is to be able to pivot across to small molecule CIDs. The steps to do this are shown below, highlighted in yellow.
The result below, transformed into a PubChem compound query, now retrieves all CIDs that include GtoPdb approved SIDs
Thus GtoPdb contains 1586 approved drug CIDs. Note that not all these are strictly “small molecules as we can see from the Mw ranking below where the top-ten are above 5000 Mw
The latest release of the IUPHAR/BPS Guide to Pharmacology database was made on 13th December 2022. This database release is version 2022.4, and is the fourth release this year. GtoPdb now contains:
Our PostgreSQL version has been update to 12.13.
This manuscript (1) reports on extensive and compelling experimental evidence and translational data from retrospective review of clinical registries, which show that pharmacological inhibition of the farnesoid X receptor (FXR) could be a novel intervention for the prevention or treatment of COVID-19. The in vitro and in vivo studies show that FXR antagonism reduces ACE2 expression, which subsequently reduces SARS-CoV-2 infection in the lung, liver and gastrointestinal tract.
Brevini T, Maes M, Webb GJ, John BV, Fuchs CD, Buescher G, Wang L, Griffiths C, Brown ML, Scott WE 3rd, Pereyra-Gerber P, Gelson WTH, Brown S, Dillon S, Muraro D, Sharp J, Neary M, Box H, Tatham L, Stewart J, Curley P, Pertinez H, Forrest S, Mlcochova P, Varankar SS, Darvish-Damavandi M, Mulcahy VL, Kuc RE, Williams TL, Heslop JA, Rossetti D, Tysoe OC, Galanakis V, Vila-Gonzalez M, Crozier TWM, Bargehr J, Sinha S, Upponi SS, Fear C, Swift L, Saeb-Parsy K, Davies SE, Wester A, Hagström H, Melum E, Clements D, Humphreys P, Herriott J, Kijak E, Cox H, Bramwell C, Valentijn A, Illingworth CJR; UK-PBC research consortium, Dahman B, Bastaich DR, Ferreira RD, Marjot T, Barnes E, Moon AM, Barritt AS 4th, Gupta RK, Baker S, Davenport AP, Corbett G, Gorgoulis VG, Buczacki SJA, Lee JH, Matheson NJ, Trauner M, Fisher AJ, Gibbs P, Butler AJ, Watson CJE, Mells GF, Dougan G, Owen A, Lohse AW, Vallier L, Sampaziotis F. FXR inhibition may protect from SARS-CoV-2 infection by reducing ACE2. Nature. 2022 Dec 5. doi: 10.1038/s41586-022-05594-0. Epub ahead of print. PMID: 36470304.
Comments by the Guide to Pharmacology Curation Team, University of Edinburgh
The latest release of the IUPHAR/BPS Guide to Pharmacology database was made on 13th October 2022. This database release is version 2022.3, and is the third release this year. The following blog post gives details of the key content updates and website changes. GtoPdb now contains:
For this release we can announce the inclusion of new ligand entries for chemical structures that have been revealed during recent ‘first disclosure’ sessions at scientific conferences. For those structures that meet our inclusion criteria, we have curated details of any clinical studies, or intended therapeutic use for these new entries, and included quantitative interaction data where we have been able to find it, as well as any source references.
2 new targets have been added, including interactions with small molecule modulators:
A host enzyme: phosphodiesterase 12 (PDE12); Two tool compounds have been added, compound 1 [PMID: 26055709] and compound 3 [PMID: 26055709], and we are awaiting disclosure of two lead compound structures that were reported in a bioRxiv preprint in Sept 2022 https://www.biorxiv.org/content/10.1101/2022.09.23.509178v1.full
A coronavirus protein: Non-structural protein 15 (aka endoribonuclease NendoU). Two allosteric inhibitors have been reported, FUZS-5 (Ligand ID 12200) and LIZA-7 (Ligand ID 12199). The docking positions of these compounds within nsp15 have been determined by X-ray crystallography https://www.biorxiv.org/content/10.1101/2022.09.26.509485v1. We have included tipiracil (8696) in the inhibitors table as it has also demonstrated some activity against SARS-CoV-2 nsp15.
As part of our continuing collaboration with AntibioticDB and GARDP we now have 351 curated antibacterial ligands in the Guide to PHARMACOLOGY.
Our chemical structure search now uses the Chemistry Development Kit (CDK) to power the similarity search. The Chemistry Development Kit (CDK) is a collection of free and open-source modular Java libraries for processing chemical information.
A new database release (version 2022.2) of the IUPHAR/BPS Guide to Pharmacology was made on 9th June 2022. This blog post gives details of the key content updates and website changes. GtoPdb now contains:
Selectin E (OID 3200) and a selective antagonist uproleselan (LID 11995): Selectin E is a vascular adhesion molecule present on the endothelial cell surface that is involved in leukocyte migration and in cancer cell trafficking (especially metastasis). Uproleselan-mediated antagonism of selectin E binding is being investigated for anti-cancer potential and has been repositioned for anti-inflammatory action in patients with severe COVID-19 pneumonia (predicted to reduce leukocyte recruitment to the inflamed lungs).
Palmitoyl-protein thioesterase 1 (OID 3199) and the inhibitor ezurpimtrostat (LID 11991): Ezurpimtrostat (GNS561) is a small molecule inhibitor of palmitoyl-protein thioesterase 1 (PPT1). It induces lysosomal dysregulation which leads to inhibition of late-stage autophagy and cell death. Antitumour and antiviral activities have been demonstrated in vitro and in vivo. Anti-SARS-CoV-2 activity is being investigated in Phase 2 clinical trial NCT04637828.
CoV Non-structural protein 14 (nsp14 OID 3198) has been added as a separate entry in the Coronavirus (CoV) proteins family to allow effective curation of nsp14 inhibitors. The methyltransferase (MTase) activity of nsp14 is required for capping of the viral RNA transcript. Inhibition of this activity should disrupt viral replication. Two experimental inhibitors have been curated, TO507 (LID 11981) and compound 25 [PMID: 35439007] (LID 11971).
We have added those ligands from the DrugHunter ‘Molecules of the month’ sets for April and May that were absent from our curated ligands. Of the total April/May combined set of 18 compounds, we had already included 5 through our own curation efforts, and added 11 as new entries. We did not add branaplam (a RNA-targeted splicing modulator for spinal muscular atrophy and/or Huntington’s disease) as it doesn’t act via a defined protein target, and we didn’t add the HIV-1 protease inhibitor ‘compound 14’ as we currently don’t include HIV proteins as ligand/drug targets.
We reviewed the compounds in current COVID-19 clinical trials (from clinicaltrials.gov) to identify agents (particularly small molecule compounds) that either didn’t exist in the GtoPdb or which needed updated curation wrt clinical potential as antivirals or treatments for symptomatic COVID-19. The results include:
In May, the FDA approved 3 drugs that are ‘first-in-class’ therapeutics. In date order of approval these are:
Congratulations to Professor Jamie Davies, Professor of Experimental Anatomy, University of Edinburgh, who has been elected to a Fellowship by The Royal Society of Edinburgh, Scotland’s National Academy.
Fellowships are awarded to individuals in academia, business and public service who are judged to be eminently distinguished in their subject.
Jamie has led the IUPHAR/BPS Guide to PHARMACOLOGY database since 2014, having taken over from Professor Tony Harmar (also a Fellow of the Society), and has been instrumental in the launch of IUPHAR Guide to IMMUNOPHARMACOLOGY and IUPHAR/MMV Guide to Malaria PHARMACOLOGY.
Human ageing is just disintegration, but absolutely precise, linked to loss of VO2 max, which is why COVID is age-related.
A new study (1) shows that the decline in human performance with age at 5000 m, an athletic event requiring high VO2 max (oxygen use), is remarkably precise, and unavoidable, and related to entropy, even at an individual level. Women and men show an identical age-related decline, up to ~100 years old. The precision of the decline shows the limitations for therapy of aging. Mortality incidence for COVID19 shows a similar relationship. The authors propose that initial VO2 max has a critical role in COVID sensitivity because of the direct relationship of disease severity with oxygen use, and the parallel decline in aging: mitochondria are targeted by viruses. The highest human recorded VO2 max is 96, but is only 13 in nursing home residents, where metabolic capacity is massively reduced.
Humans, with a long active life, may therefore have already optimised ageing and metabolism (2) compared to short-lived animals which are used in studies of anti-ageing drugs.
As the results involve the very best age-related performances of humanity (by definition healthy), and there is absolute mathematical precision in the decline, there is no room for improvement, so drugs to slow ageing (apart from offsetting specific ageing-related diseases) may well remain a mirage. The results of the study are compatible with decline due to entropy, just a general disintegration, with different diseases developing by chance, heredity or the environment in a disintegrating body.
Funding could be better spent on ensuring a humane and dignified exit at the end of life when metabolism breaks down completely. Nevertheless, exercise (such as 5000M parkruns) has been previously shown to extend active life by ~7 years (3).
Comments by the Guide to Pharmacology Curation Team, University of Edinburgh
SARS-CoV-2 gains entry into the body mainly via the lungs. After entering the blood, the virus rapidly multiplies to infect nearby cells. Endothelial cells line every blood vessel and have a surface area similar to a tennis court. These cells are especially vulnerable to virus infection as they express the the virus entry receptor, ACE2. Endothelial cells are also a source of endothelin-1 (ET-1), which is released in response to injury to cause long lasting vasoconstriction, reducing oxygen supply to vital organs. In the lungs, this can cause breathing difficulty, which in severe cases requires patients to be given oxygen and put on a ventilator.
The study compared ET-1 in the plasma of patients infected with COVID-19 who were hospitalised during the first wave of the pandemic, including those placed on ventilators (1). Significantly higher levels of ET-1 were measured in these patients, compared with infected individuals with mild or no symptoms. The release of multiple copies of the virus following infection is known to damage endothelial cells and the results show stored ET-1 is also released into the bloodstream.
Currently, medicines that block the detrimental actions of ET-1 are used to treat rare forms of lung disease that share some of the clinical features of COVID-19 infection. The results provide evidence to support testing these medicines in clinical trials to see if they would benefit the most severely affected patients. The virus continues to evolve and in addition to vaccines and bespoke antivirals, that may be susceptible to the development of resistance, it is important to develop alternative ways to block the harmful actions of the virus. By targeting the damaging actions of SARS-CoV-2 on the host cells, medicines are likely to remain effective irrespective of evolving mutations in the virus.
Comments by the Guide to Pharmacology Curation Team, University of Edinburgh
We are pleased to announce that the first IUPHAR/BPS Guide to Pharmacology of 2022 was made on 31st March 2022. database. This release is version 2022.1 and this blog post gives details of the key content updates and website changes. GtoPdb now contains:
The Guide to Pharmacology coronavirus information page continues to be updated on a regular basis to capture the latest pharmacological strategies under investigation to mitigate against COVID-19.
Notable additions and updates for this period include:
Baricitinib – In March 2022, data reported from the largest of the baricitinib studies (part of the University of Oxford-led RECOVERY trial) indicated that it provided clinical benefit in hospitalised COVID-19 patients, including in those already receiving other standard care immunomodulatory treatments (e.g.dexamethasone, tocilizumab) or the antiviral drug remdesivir. This made baricitinib the 4th effective COVID-19 therapy to be identified by the RECOVERY trial.
N-0385 – A serine protease inhibitor, that was originally reported as a matriptase inhibitor. More recently revealed to inhibit SARS-CoV-2 infection via inhibition of TMPRSS2 (the host protease hijacked by the coronavirus to cleave spike and facilitate entry into host cells), thus functioning as an ‘entry inhibitor’. The Nature article (DOI: 10.1038/s41586-022-04661-w) proposes prophylactic and therapeutic potential, as well as pan-coronavirus efficacy that is independent of virus strain. May have applications in combination with antivirals with other MMOAs e.g. Mpro (nirmatrelvir) or RdRp (e.g. molnupiravir , remdesivir) inhibitors.
2021.4 update we have added 131 new ligands. We have also made updates to ~38 existing ligand comments and ~28 clinical use comments. An additional 10 existing ligand have now been tagged as approved drugs, bringing the total approved drugs in GtoPdb to 1,730.
All new ligands have been manually curated with chemical or peptide structures, links to external resources, general comments, notes on clinical development where appropriate, target interaction data and patent pointers where available. Two particular sets are from recent collaborative blogposts. The first consisted of six DrugHunter compounds from Feb 2022. The second set of six was the popular ACS First Disclosures from their March spring meeting (see BLU945 below). For most of these fresh drug discovery ligands (some of which are patent-only and not yet published) we will be the first source to provide name-to-structure matches both in our own database and in PubChem (including PubChem < > PubMed links).
A few new interesting ligands are:
Whilst no new targets have been added to the database for this release we want to draw attention to the set of novel protein targets for drugs that were approved by the FDA (US), EMA (EU) and/or PMDA (Japan) in 2021. 14 targets of the 2021 approved drugs set are novel, and 9 of these 14 are indicated for rare diseases*. Links to our pages for these targets and drugs (where available) are provided in the table below :
These are the recent advancements made in the GtoMPdb for this database release:
The Antimalarial targets family and the Antimalarial ligands family have been updated, giving a total of 40 P. falciparum (3D7) targets and 134 ligands tagged as antimalarial in the database. Work has continued on the process of adding subfamilies to the Antimalarial targets family.
ACE2 (Guide to Pharmacology Target id: 1614) normally functions as an enzyme metabolising peptides that regulate the cardiovascular system. As is well known though, it has recently gained additional scientific fame by also acting as a receptor for SARS-CoV-2. This study [1] compared the distribution in human tissue of ACE2 versus deltaACE2, a recently discovered truncated isoform that has lost the spike binding sites for SARS-CoV-2, and is predicted to confer reduced susceptibility to viral infection. ACE2 was highly expressed in human lung, kidney, heart, liver and vasculature. However, deltaACE2 was the predominant protein alternative initiation variant in lung airway and liver bile duct epithelia, which may be a contributing factor to variation in response to the virus. A diagrammatic relationships of the full length and dACE is shown below.
The discovery of this novel short dACE2 variant in two independent reports may have important implications in COVID-19 research. It is enzymatically inactive, and the absence of the spike binding domains of the full-length ACE2 suggests dACE2 does not bind SARS-CoV-2 spike protein. It is still unclear what the precise effects of such an alternative form are in both normal physiology and in relation to SARS-CoV-2 infection and Long COVID, but the ratio of dACE2 versus the full-length protein may be a contributing factor in the wide inter-individual variation in response to COVID-19
Contributed by Christopher Southan, ORCID 0000-0001-9580-0446, Data Science, Medicines Discovery Catapult and Honorary Fellow, University of Edinburgh
The 2021.4 release of the IUPHAR Guide to Pharmacology was made on 14th December 2021. This blog post gives details of the key content updates and website change The 2021.4 release contains:
The Guide to Pharmacology coronavirus information page continues to be updated on a regular basis to capture the latest pharmacological strategies under investigation to mitigate against COVID-19.
Updates have most notably been made to the comments for molnupiravir (EIDD-2801, MK-4482). This followed its MHRA approval on 4th Nov 2021 as a treatment or recently diagnosed SARS-CoV-2 infection in non-hospitalised individuals who have at least one risk factor for developing severe illness.
Also add to the ligand list is PF-07321332, an oral SARS-CoV-2 3CL protease (Mpro) inhibitor, and clinical lead compound from Pfizer. It was progressed to clinical trial to determine safety and efficacy as a treatment for COVID-19. Phase 1 (NCT04756531) results were published in Science, in early November 2021 DOI: 10.1126/science.abl4784. A few days after the Phase 1 results were published, Pfizer announced that interim analysis from their Phase 3 EPIC-HR study (NCT04960202), indicated that PF-07321332 (in combination with ritonavir, as a formulation with the trade name Paxlovid) reduced the risk of hospitalisation or death by almost 90% in patients who were at high risk of progressing to severe illness, and who were treated within 3 days of symptom onset https://bit.ly/3wxA5of.
OAS1 (HGNC:8086; UniProt: P00973 ), an interferon-induced gene that is part of the innate antiviral defence system within host cells, has been added to the target table.
Since our 2021.3 update we have added over ~110 new ligands. These have all been manually curated with chemical or peptide structures, links to external resources, general comments, notes on clinical development where appropriate, and target interaction data where this is available.
We have also made updates to ~46 existing ligand comments and ~52 clinical use comments. An additional 18 existing ligand have now been tagged as approved drugs, bringing the total approved drugs in GtoPdb to 1,716.
5 new targets have been added to the database since the last release. These include HtrA serine peptidase 1, the target of galegenimab (Ph 2 geographic atrophy); heat shock protein family A (Hsp70) member 5, a potential therapeutic target for viral infections (incl. SARS-CoV-2), chemoresistant cancers and inflammation; coagulation factor III, tissue factor , the target of tisotumab vedotin (approved, oncology);clusterin : the target of sotevtamab (Ph 2 immunomodulator, antineoplastic); and transferrin receptor (CD71), which is exploited to improve site-specific drug delivery, including drug delivery across the blood-brain barrier e.g. pabinafusp alfa, lepunafusp alfa that deliver enzymes that are deficient in lysosomal storage disorders (both approved in Japan).
Screenshot of the new GtoPdb home page. The site search is now more prominent, alongside the more advance search tools.
These are the recent advancements made in the GtoMPdb for this database release:
It is with great pleasure that we can announce that the IUPHAR/BPS Guide to PHARMACOLOGY has been given a hidden REF award in the category ‘applications of research’.
Guide to Pharmacology hidden REF certificate
The hidden Ref (https://hidden-ref.org) is a national ‘competition’, supported by publishers, learned societies etc. (https://hidden-ref.org/supporters/) , designed to celebrate and recognise the range of important research achievements that may not fit neatly into a REF submission.
“The ways in which the research impact is judged overlooks many of the people who are vital to the success of research. It’s only by recognising everyone who is vital to the conduct of research that we will create an environment in which to advance it.”
We are of course very grateful to receive this award, and our thanks go to the hidden REF committees.
Being recognised in this way is a testament to the hard work of the entire Guide to PHARMACOLOGY team, both past and present, who’s vision and dedication has provided the research community with such an invaluable resource.
The award ceremony is available to view and you can watch the specific announcement of the GtoPdb award in the following video (starts ~10 mins in):
More information on the hidden REF is provide in this video:
The new release of the IUPHAR Guide to Pharmacology was made on 2nd September 2021 – this is version 2021.3. This blog post gives details of the key content updates and website changes.
The 2021.3 release contains:
The Guide to Pharmacology coronavirus information page continues to be updated on a regular basis to capture the latest pharmacological strategies under investigation to mitigate against COVID-19.
Most recently we have added the DX600 experimental peptide, which has been shown to prevent pseudotyped SARS-CoV-2 from entering heart cells (Williams et al., Commun. Biol, 2021). Also added are two monoclonal antibody cocktails. The first, AZD7442, is a combination of cilgavimab and tixagevimab, which reach phase 3 clinical evalutaion, and is proposed to have therapeutic potential in treating individuals who respond poorly to vaccination. The second, Ronapreve, is a combination of imdevimab and casirivimab, aimed at blocking SARS-CoV-2 entry into cells it was approved by the UK’s MHRA in August 2021 having previous been approved by both the FDA and Japanese Ministry of Health.
Since our 2021.2 update we have added over ~120 new ligands. These have all been manually curated with chemical or peptide structures, links to external resources, general comments, notes on clinical development where appropriate, and target interaction data where this is available.
We have also made updates to over 170 existing ligand comments and 102 bioactivity comments. An additional 8 existing ligand have now been tagged as approved drugs, bringing the total approved drugs in GtoPdb to 1,688.
These are the recent advancements made in the GtoMPdb for this database release:
The FDA approval of imatinib in 2001 was a breakthrough in molecularly targeted cancer therapy and heralded the emergence of kinase inhibitors as a key drug class in the oncology area and beyond. Continued advances in the molecular understanding of cancer, multiple approaches to drug design, and the increasing number of resolved kinase structures (1) have facilitated the development of kinase inhibitors with improved potency, selectivity, and efficacy.
Two new analyses published in Nature Reviews Drug Discovery present the historical development of kinase inhibitors as well as the current outlook on kinase drug discovery. Cohen et al. (1) present a detailed study on FDA-approved small molecular weight kinase inhibitors titled Kinase drug discovery 20 years after imatinib: progress and future directions. The analysis encompasses the pivotal events in kinase inhibitor development and remarkable progress made over the past 20 years in improving the potency and specificity of small molecule kinase inhibitors (SMKIs) and the kinase pathways targeted in drug discovery. Importantly, the development of drug resistance to kinase inhibitors and how these challenges are being met in the future of kinase drug discovery are discussed.
The new analysis Trends in kinase drug discovery: targets, indications and inhibitor design published this month by Attwood et al. (2) analyses the landscape of approved and investigational therapies targeting kinases and trends within it, including the most popular targets of kinase inhibitors and their expanding range of indications. Furthermore, strategies for kinase inhibitor design, including the development of allosteric and covalent inhibitors, bifunctional inhibitors, and chemical degraders are discussed. Since the approval of fasudil in 1995, the number of approved kinase inhibitors worldwide as increased to 98 drugs, of which 71 are SMKIs and 10 monoclonal antibodies approved by the FDA. Remarkably, the number of FDA-approved SMKIs has more than doubled in the past five years and they constitute ~15% of all novel drug approvals by the FDA. While oncology is still the predominant area for their application, there have been important approvals for indications such as rheumatoid arthritis, and one-third of the SMKIs in clinical development address disorders beyond oncology. Nearly 600 investigational kinase-targeting agents that were registered in ClinicalTrials.gov were included this analysis, consisting of 475 novel SMKIs and 124 biological agents. Information on clinical trials of SMKIs reveals that ~110 novel kinases are currently being explored as targets, which together with the ~45 targets of approved kinase inhibitors represent only ~30% of the human kinome (4), indicating that there are still substantial unexplored opportunities for this drug class.
In summary, although there have been substantial advances in kinase drug discovery, there are still many challenges and opportunities in this field. The potential for developing novel types of kinase inhibitors is large and it is expected that this will continue to be a major area of growth in the next 20 years.
Comments by Misty Attwood and Helgi Schiöth, University of Uppsala, Uppsala, Sweden. Helgi Schiöth is Chair for NC-IUPHAR Subcommitees for Melanocortin receptors and Prolactin-releasing peptide receptor. Twitter: @FunctPharm
The new release of the IUPHAR Guide to Pharmacology was made on 25th June 2021 – this is version 2020.2. This blog post gives details of the key content updates and website changes.
The 2021.2 release contains:
Since our 2021.2 update we have added over 70 new ligands. These have all been manually curated with chemical or peptide structures, links to external resources, general comments, notes on clinical development where appropriate, and target interaction data where this is available. We have also made update to over 40 existing ligand comments and clinical use summaries. Bioactivity comments have been updated on 10 ligands and 16 existing ligand have now been tagged as approved drugs.
These are the recent advancements made in the GtoMPdb for this database release:
The Guide to Pharmacology coronavirus information page continues to be updated on a regular basis to capture the latest pharmacological strategies under investigation to mitigate against COVID-19.
Here are the details for the first release in 2021 of the Guide to PHARMACOLOGY database, version 2021.1. This is the last release before we move forward with the process of preparing the next edition of the Concise Guide to Pharmacology (2021/22). As such, the release contains many updates specifically with the CGTP in mind.
The 2021.1 release contains:
The Guide to Pharmacology coronavirus information page continues to be updated on a regular basis to capture the latest pharmacological strategies under investigation to mitigate against COVID-19.
The peptide GM-CSF has been added to the targets lists as this has been indicated as a principal marker of sever COVID-19 immunopathology. Ligands added in include ensovibep (MP0420), emovododstat & MK-7110. Updates made to comments for sarliumab and tocilizumab.
There have been some fairly significant updates in this release as a precursor to preparing the 2021/22 edition of the Concise Guide to Pharmacology.
A new family of enzymes, Peptidyl-prolyl cis/trans isomerases (PPIases), has been added. This includes some of the best understood PPIases, in particular those that are therapeutic targets (current or under investigation). We have added several pharmacological modulators that can be used to investigate their biological roles, as well as relevant clinical candidate compounds.
Ten of our ligands have been updated to reflect their approval by the FDA in the first quarter of 2021: vericiguat, cabotegravir, voclosporin, tepotinib, umbralisib, evinacumab-dgnb, trilaciclib, casimersen, serdexmethylphenidate and tivozanib.
Since our 2020.5 update we have added 220 new ligands. These have all been manually curated with chemical or peptide structures, links to external resources, general comments, notes on clinical development where appropriate, and target interaction data where this is available.
Many of our target family summary pages have been reviewed by our expert committees, and revised in preparation for generating the next edition of the Concise Guide to PHARMACOLOGY (2021/22 ed).
These are the recent advancements made in the GtoMPdb for this database release:
[画像:screenshot-2021年02月25日-at-18.39.57]Figure 1. The Antimalarial targets family page illustrating the new subfamily classification (highlighted in magenta).
These are some of the ligands under curation (pre-release) in the Guide to Pharmacology. We expect them to be available on the website at the next database release (2021.1).
Ligand ID: 11395
Names: MK-7110
Comment: MK-7110 (originally OncoImmune’s CD24Fc) is a synthetic protein that fuses the nonpolymorphic regions of CD24 (which is an agonist of Siglec-10) to the Fc region of human IgG1 [PubMed ID:19264983]. It.is being investigated as a modulator of the innate immune system, originally as an intervention to ameliorate graft-versus-host disease in leukemia patients receiving stem cell transplants, or autoimmune diseases. The peptide’s sequence was extracted from OncoImmune’s patent US20130231464A1 the first 30 amino acids are the CD24 region and the remainder belong to the IgG1 Fc domain.
Coronavirus relevance: A clinical stage synthetic fusion protein and CD24 mimetic, that enhances CD24/SIGLEC10 suppression of DAMP-triggered activation of the immune response and associated tissue damage. Repositioned to combat COVID-19 inflammation (currently Phase 3).
Ligand ID: 11421
Names: emvododstat
Comment: Emvododstat (PTC299) is an orally bioavailable inhibitor of dihydroorotate dehydrogenase (DHODH) that was developed by PTC Therapeutics for anti-cancer potential. It inhibits de novo pyrimidine nucleotide (UMP) biosynthesis and exhibits broad activity against leukemia in vitro and in vivo.
Coronavirus relevance: Emvododstat inhibits replication of SARS-CoV-2 in vitro (EC50 2.0-31.6 nM) and other RNA viruses. It blocks production of inflammatory cytokines in infected cell cultures.
Ligand ID: 11470
Names: ensovibep (MP0420)
Comment: MP0420 is a DARPin that binds to the SARS-CoV-2 spike glycoprotein, and offers virus neutralising potential. It is being progressed to clinical trial by Molecular Partners and Novartis. DARPins are protein-based therapeutics, with the high selectivity and affinity of antibodies, but are around 10% of the size. They are easier to manufacture than whole antibodies, and may be suitable for subcutaneous administration rather than infusion. Molecular Partners have developed two multi-valent DARPins that are designed to engage multiple sites of the SARS-CoV-2 spike glycoprotein. Novartis and Molecular Partners are working together on both MP0420 and MP0423 for COVID-19.
The GtoPdb team need no convincing about the importance of eliminating equivocality in gene and gene product names, as they are used in the pharmacology, chemical biology and drug discovery literature. Crucially, this also applies to their inclusion in curated database bioactivity records such as GtoPdb. We were thus particularly pleased to see “Standardizing gene product nomenclature” that appeared recently in PNAS [1]. This included not only an honourable mention of the Nomenclature Committee of the International Union of Basic and Clinical Pharmacology (NC-IUPHAR) but also the citation of our most recent Concise Guide Introduction [2].
The collaboration between NC-IUPHAR and HGNC for standardising the nomenclature of pharmacological targets goes back well over a decade. This has been (and continues to be) an iterative process whereby NC (after extensive consultation) recommends naming schema for certain target classes that are different to those approved by HGNC. This is typically done on the basis of well established publication usage in the pharmacological community. In many cases these older protein family names predate completion of the human genome. This can be illustrated for the case of the Voltage-gated calcium channels as shown in the GtoPdb family page below.
As we can see in the page for Cav1.1 curation adds a full set of synonyms including CACNA1S and “calcium voltage-gated channel subunit alpha1 S” as approved HGNC Symbol and name, respectively. Below we can see the reciprocal cross-pointing between HGNC and GtoPdb/NC-IUPHAR.
We have commented some time ago on ambiguity issues arising from the necessity of curatorial resolution for authors’ descriptions of key entities to standardized identifiers, including mapping between ligands and target protein names [3]. We consequently hope both papers will contribute to greater use of both HGNC and NC-IUPHAR nomenclature in the pharmacology literature.
Comments by Chris Southan, Fellow of the University of Edinburgh, Owner of TW2Informatics, Chair of NC-IUPHAR Subcommitees for Proteases and Drug Targets and Chemistry (DRUTACS).
[1] Kenji Fujiyoshi, Elspeth A. Bruford, Pawel Mroz, Cynthe L. Sims, Timothy J. O’Leary, Anthony W. I. Lo, Neng Chen, Nimesh R. Patel, Keyur Pravinchandra Patel, Barbara Seliger, Mingyang Song, Federico A. Monzon, Alexis B. Carter, Margaret L. Gulley, Susan M. Mockus, Thuy L. Phung, Harriet Feilotter, Heather E. Williams, and Shuji Ogino (2021) Opinion: Standardizing gene product nomenclature-a call to action, Proc Natl Acad Sci U S A Jan 19;118(3) doi: 10.1073/pnas.2025207118, PMID: 33408252.
[2] Stephen P H Alexander, Eamonn Kelly, Alistair Mathie, John A Peters, Emma L Veale, Jane F Armstrong, Elena Faccenda, Simon D Harding, Adam J Pawson, Joanna L Sharman, Christopher Southan, O Peter Buneman, John A Cidlowski, Arthur Christopoulos, Anthony P Davenport, Doriano Fabbro , Michael Spedding, Jörg Striessnig , Jamie A Davies, CGTP Collaborators (2019) The Concise Guide to Pharmacology 2019/20: Introduction and Other Protein Targets, Br J Pharmacol. Dec; 176 (Suppl 1): S1–S20, PMID: 31710719.
[3] Christopher Southan 1, Joanna L Sharman 1, Elena Faccenda 1, Adam J Pawson 1, Simon D Harding 1, Jamie A Davies 1 (2018) Challenges of Connecting Chemistry to Pharmacology: Perspectives from Curating the IUPHAR/BPS Guide to PHARMACOLOGY ACS Omega, Jul 31;3(7):8408-8420, PMID: 30087946.
The BPS Pharmacology 2020 Meeting is being held virtually this year, but this hasn’t diminished Guide to Pharmacology presence.
On Monday 14th, Dr. Simon Harding presented our poster on ‘Expansion for anti-malarial, antibiotics and COVID-19’. Click to view poster.
On Tuesday 15th, Dr. Chris Southan gave on oral presentation on Curating SARS-CoV-2 viral targets for the IUPHAR/BPS Guide to Pharmacology’. The session is currently available ‘on-demand’ at this link. These are available to register attendees for one month.
Chris also presented a poster on ‘SARS-CoV-2/COVID-19 pharmacological roadmap: strategy for curating and updating drug targets in the Guide to Pharmacology Coronavirus Information page’. Click to view poster.
Follow this link to view the GtoPdb Coronavirus Information Page.
We are pleased to announce the latest release of the Guide to PHARMACOLOGY database, version 2020.5. This is the last planned release for 2020.
The 2020.5 release contains:
The Guide to Pharmacology coronavirus information page continues to be updated on a regular basis to capture the latest pharmacological strategies under investigation to mitigate against COVID-19.
We have curated some of the entities from the WHO list of COVID-related therapeutics that was released at the end of October: alunacedase alfa, eclitasertib, enpatoran, molnupiravir, nezulcitinib, subasumstat.
There have been updates to the S1P and LPA receptor families, the bradykinin receptors family, and the S1P turnover enzymes. The P-type ATPases have been expanded and reorganised.
A new family of transporters has been added, SLC66 Lysosomal amino acid transporters, and cyclic GMP-AMP synthase (cGAS) has been added as a new immunopharmacology target.
These are the recent advancements made in the GtoMPdb for this database release:
The latest release of the Guide to PHARMACOLOGY database, version 2020.4, has now been made.
The Guide to Pharmacology coronavirus information page continues to be updated on a regular basis to capture the latest pharmacological strategies under investigation to mitigate against COVID-19.
GtoPdb curates Coronavirus (CoV) proteins under viral protein targets in our hierarchy. This family contains 15 CoV proteins. Updates in this release to the CoV 3C-like (main) protease include outlinks to CHEMBL (CHEMBL3927) and UniProt (P0C6U8); the addition of 4 further 3D structures (3VB3, 2GX4, 6LZE and 6XHL) and and additional 19 curated ligand interactions.
We’ve added some new targets proteins along with associated pharmacological ligands. These include the following five, that are all oncology targets:
Also added is C-type lectin receptor CLEC4C, an anti-inflammatory target, and N-acetyltransferase 8 like (ANAT), a target for the treatment of Canavan disease which is a rare aspartoacylase deficiency-mediated neurodegenerative condition.
The list of WHO essential medicines has been expanded with the inclusion of many antibiotics and antivirals.
Overall this release sees updates made to targets across 35 targets families. These are listed below with links to family page in the database:
GPCRs
Adenosine receptors
Bradykinin receptors
Calcium-sensing receptor
Complement peptide receptors
Dopamine receptors
Gonadotrophin-releasing hormone receptors
Hydroxycarboxylic acid receptors
Leukotriene receptors
Melanocortin receptors
Melatonin receptors
Motilin receptor
Neuropeptide S receptor
Neurotensin receptors
Prolactin-releasing peptide receptor
QRFP receptor
Ion-Channels
Glycine receptors
P2X receptors
Voltage-gated potassium channels
Voltage-gated proton channel
Catalytic Receptors
Nitric oxide (NO)-sensitive (soluble) guanylyl cyclase
Transmembrane guanylyl cyclases
Tumour necrosis factor (TNF) receptor family
Enzymes
N-Acylethanolamine turnover
2-Acylglycerol ester turnover
Sphingosine 1-phosphate lyase
Sphingosine 1-phosphate phosphatase
Sphingosine 1-phosphate turnover
Sphingosine kinase
Transporters
Neutral amino acid transporter subfamily
SLC14 family of facilitative urea transporters
SLC28 family
SLC29 family
SLC51 family of steroid-derived molecule transporters
SLC8 family of sodium/calcium exchangers
Other Proteins
R7 family of RGS proteins
These are the recent advancements made in the GtoMPdb for this database release:
Historically, the main classes of drug targets have been receptors, enzymes, ion channels and transporters which are primarily targeted by small molecules. However, advances in molecular biology, genomics, and pharmacology have facilitated the development of different therapeutic modalities which in turn have broadened the types of drug targets. In particular, soluble ligands have growing interest as targets and now constitute 10% of novel targets in clinical trials and are currently the third-largest target class of therapeutic agents targeting human protein products, after enzymes and receptors [1].
A new analysis of ligand-targeting [2], illustrates the different classes of ligand-targeting drugs and targets as well as analysing the success of the different technology platforms. This emerging class of drug targets is very dynamic with 291 agents that target 99 unique ligands reaching clinical development from 1992 to 2020. In the last five years, the number of ligand-targeting agents which are FDA-approved has doubled to 34, while the number of clinically validated ligand targets has doubled to 22. Therapeutic antibodies are the most common class of both approved and investigative ligand-targeting agents, followed by decoy receptors. Several technology platforms are successfully being developed, enhancing the opportunities of drug development using antibody fragments, single-domain antibodies, aptamers, spiegelmers, engineered protein scaffolds, gene therapy, therapeutic vaccines and oligonucleotides. There is an increase in the number of agents that target more than one ligand, as well as the number of unique combinations of ligands. Advances in engineering technology and the ability to effectively and safely target more than one ligand has combined with increased knowledge of disease pathways to effectively utilize bispecific antibodies and decoy receptors, amongst others, to selectively disrupt multiple biological pathways by blocking two (or more) related or unrelated ligands. Cytokines and growth factors are the predominant types of targeted ligands (70%), which is consistent with the three major disease groups being treated by both approved and investigational agents: inflammation and autoimmune diseases, cancer, and ophthalmological diseases.
Several factors contribute to the increasing importance of soluble ligands as drug targets, including the growing understanding of the role of the immune system in many diseases in conjunction with the tractability of cytokines as therapeutic agents, and the relative accessibility of ligands in comparison to their receptors to therapeutic agents. Given the increasing body of evidence that inflammation is involved in many diseases beyond typical inflammation disorders and the position of ligand-targeting drugs at the forefront of anti-inflammatory therapies, there will be continued interest in this class of agents. Furthermore, there is vast territory to explore in the ligand target landscape, as there are almost 600 endogenous human peptides according to IUPHAR/BPS Guide to PHARMACOLOGY (https://www.guidetopharmacology.org) with approximately 150 of these identified as having specifically curated immunopharmacological data associated with them (IUPHAR Guide to IMMUNOPHARMACOLOGY: https://www.guidetoimmunopharmacology.org/immuno/).
In conclusion, ligands as a type of drug target now merit consideration as a distinct and expanding group of targets for a range of therapeutic modalities that can exert their effects through single targets or selected combinations.
Comments by Misty Attwood and Helgi Schiöth, University of Uppsala, Uppsala, Sweden. Helgi Schiöth is Chair for NC-IUPHAR Subcommitees for Melanocortin receptors and Prolactin-releasing peptide receptor. Twitter: @FunctPharm
These are some of the ligands under curation (pre-release) in the Guide to Pharmacology. We expect them to be available on the website at the next database release (2020.4).
Ligand ID: 11132
Name: otilimab
Comment: Otilimab (MOR103, GSK3196165) is a clinical stage fully human IgG1λ antibody that targets the granulocyte-macrophage colony-stimulating factor (GM-CSF; CSF2). It is being investigated as a therapeutic for inflammatory diseases. MOR103 was developed using MorphoSys’ HuCAL® technology, and has been fully out-licensed otilimab to GSK. MOR103 is claimed as MOR-04357 in patent WO2006122797A2 (by BLAST peptide sequence matching).
Ligand ID: 11139
Name: favipiravir
[画像:11139]
Comment: Favipiravir (T-705) is an orally delivered, guanine (purine) analogue antiviral drug (cf. remdesivir which is administered i.v.). It targets viral RNA-dependent RNA polymerase (RdRP) of RNA viruses and since the catalytic domain of RdRP is well conserved across species, has a broad-spectrum of activity; although Furuta et al. (2002) reported that actvity was weak against non-influenza virus RNA viruses [4]. Favipiravir was originally identified through a chemical library screen against influenza virus RdRP [3]. Chemically, it is a prodrug. In human cells it undergoes phosphoribosylation and phosphorylation to its active form, favipiravir-ribofuranosyl-5′-triphosphate (F-RTP). F-RTP is bound by the RdRP, but it blocks enzyme activity and so terminates chain elongation [5-6].
IUPAC Name: 5-fluoro-2-oxo-1H-pyrazine-3-carboxamide
It is now clear that ligand-gated ion channels (LGICs) are not "stand alone" functional units, but form complexes with other components, including scaffolding proteins, regulatory proteins and enzymes. Besides their important physiological roles, these modulating proteins are also potential targets for drug discovery. P2X receptors are LGICs for which ATP is the endogenous agonist. Seven P2X subunits have been identified and they form trimers to produce at least twelve different receptor subtypes. Here, the authors combined a genome-wide open reading frame (ORF) collection with high-throughput functional screening, to search for P2X receptor modulators.
They first created a HEK-293 cell line that stably expressed human P2X2 plus P2X3 receptors, then one-by-one, co-expressed each of 17,284 non-redundant ORFs, which represents 90% of the known human protein-coding genes. They then compared the rise in intracellular [Ca2+] induced by the P2X3 agonist α,β-meATP, using a composite score of four measurements; 1) baseline, 2) peak and 3) steady-state [Ca2+], along with 4) time-course of the decay of the peak. The highest scoring ORFs were then co-expressed with P2X3 receptors in Xenopus laevis oocytes and their effects on ATP-induced ion currents determined. This process led to TMEM163 being identified as a modulator of P2X3 receptors.
TMEM163 comprises 289 amino acids, with a molecular weight of 31 kDa and predicted secondary structure of six transmembrane-spanning domains with intracellular NH2– and COOH-termini. When expressed on its own, TMEM163 had no ATP-dependent activity and when co-expressed, had no effect on the activity of AMPA, kainate, muscarinic or P2X2 receptors, nor on endogenous P2Y receptors in HEK-293 cells. It did, however, potentiate ATP-evoked currents mediated by P2X1 and P2X4 receptors and conversely, inhibited currents carried by P2X7 receptors. Thus TMEM163 appears to selectively modulate P2X receptor activity, but in a subtype-specific manner.
More detailed analysis of its actions showed that TMEM163 potentiated P2X3 receptors by shifting the ATP concentration-response curve (CRC) to the left, with a five-fold decrease in the EC50 and slowing the time-course of current decay. Surprisingly, it also substantially reduced the surface expression of P2X3 receptors. In contrast, the inhibitory effects on P2X7 receptors were associated with a decrease in both the potency of ATP and surface expression of the receptor.
Next, the expression and effects of native TM163 was investigated in mice. In situ hybridisation and an anti-TM163 antibody showed TMEM163 to be present in primary cultures of mouse cerebellar neurones. Infection with a TMEM163 shRNA reduced protein expression by almost half and the peak amplitude of ATP-induced ion currents by a third. The effect on P2X3 receptor protein levels was not reported, however. To study its role in vivo, TMEM163 was knocked-out using CRISPR/Cas9 and pain-associated behaviour induced by ATP injection into the hind-paw studied. There was no difference in paw lifting between wild-type and knock-out mice, but freezing behaviour was reduced by about 50%. Notably, and in contrast to the data obtained using recombinant TMEM163, the total amount of P2X3 protein in dorsal root ganglion was unchanged. The ATP CRC was, however, shifted about five-fold to the right by TMEM163 knock-out, consistent with the leftwards shift seen using the recombinant proteins.
By showing that that TMEM163 selectively modulates P2X receptor activity and in a subtype-specific manner, this study greatly improves our understanding of how native P2X receptors function. As such, TMEM163 is potentially a new target for drug discovery.
Comments by Charles Kennedy , Strathclyde Institute of Pharmacy and Biomedical Sciences, University of Strathclyde, Glasgow, UK. Chair for NC-IUPHAR Subcommitee for P2X receptors.
The Guide to Pharmacology (GtoPdb) currently contains data on over 10,000 different ligands which are categorised into an number of different groups or classifications. The ligand list page (https://www.guidetopharmacology.org/GRAC/LigandListForward?type=WHO-essential&database=all) provides a directory of all the ligands described in the database, divided into subsets according to chemical category.
We have recently added a WHO Essential Medicine subset, which contains ligands in GtoPdb that are also in the latest WHO Model List of Essential Medicines (21st list, 2019) (https://www.who.int/medicines/publications/essentialmedicines/en/). This totals 224 ligands in our 2020.3 release.
Selecting the WHO tab from the ligand list page shows this subset (Figure 1). Users can also download this set of ligands (as csv file) by clicking on the link in the top-right.
Figure 1. GtoPdb ligand list page showing WHO subset. https://www.guidetopharmacology.org/GRAC/LigandListForward?type=WHO-essential&database=all
Accessing WHO tagged GtoPdb compound in PubChem
GtoPdb regularly uploads its set of compounds to PubChem, which provides all GtoPdb ligands with PubChem Substance Identifiers (SIDs), which are in turn mapped (by PubChem) to PubChem Compound Identifiers (CIDs). As part of our PubChem upload process we have developed our curatorial tagging within the depositor comment sections in the substance records (SIDs). This means users are able to make domain-specific queries, to be executed from both the PubChem Substance (SID) and PubChem Compound (CID) interfaces, by using an advanced search of ‘comments’ fields.
We have now added a WHO tag to the comments fields in our PubChem upload (see example for cholorquine, Figure 2). [画像:chloroquine2]
Figure 2. Depositor comment for Chloroquine (PubChem Substance ID 178102177) showing GtoPdb curatorial tagging to mark substance as included in WHO Essential Medicines List. https://pubchem.ncbi.nlm.nih.gov/substance/178102177#section=Depositor-Comments
Therefore it is now possible, through the query below to easily access the WHO Essential Medicines in GtoPdb through PubChem.
Figure 3. Search result in PubChem for IUPHAR/BPS Guide to PHARMACOLOGY substances that include ‘gtopdb_who’ in the comments. https://www.ncbi.nlm.nih.gov/pcsubstance/?term=(%22iuphar%2Fbps+guide+to+pharmacology%22%5BSourceName%5D)+AND+%22gtopdb+who%22%5BComment%5D
The latest release of the Guide to PHARMACOLOGY database, version 2020.3, has now been made. A large focus of our curation over the last couple of month since our 2020.2 release We are pleased to have been able to make an expeditious database release (version 2020.2), following on from our last update in March 2020 in order to make public new curation specifically related to SARS-CoV-2.
The Guide to Pharmacology coronavirus information page continues to be updated on a regular basis to capture the latest pharmacological strategies under investigation to mitigate against COVID-19.
Recently published in BJP is our review that “sets out to identify opportunities for drug discovery in the treatment of COVID‐19 and in so doing, provide a rational roadmap whereby pharmacology and pharmacologists can mitigate against the global pandemic“:
Alexander SPH, Armstrong J, Davenport AP, Davies JA, Faccenda E, Harding SD, Levi-Schaffer F, Maguire JJ, Pawson AJ, Southan C, Spedding MJ. (2020). A rational roadmap for SARS‐CoV‐2/COVID‐19 pharmacotherapeutic research and development. IUPHAR Review 29 Br J Pharmacol. doi: 10.1111/bph.15094. [PMID:32358833]
Main target families with updated in this release:
GPCRs
Ion Channels
NHRs
Enzymes, Catalytic Receptors
New drug target
Acyl-CoA synthetase short chain family member 2 (ACSS2) is an emerging oncology drug target that plays a role in epithelial-mesenchymal transition (EMT) and cancer cell metastasis. Two ACSS2 inhibitor probe compounds have been curated.
AntibioticDB
As part of a collaboration with AntibioticDB (https://www.antibioticdb.com/), we have now begun to identify and tag sets of antibiotic ligands in GtoPdb. Where we have identified mappings between these and compounds in the AntibioticDB repository, we’ve put in place direct links. The newly curated set includes approved and investigational antibiotics and pre-clinical leads, as well as compounds whose development has been discontinued.
Teixobactin ligand summary page (https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=10972). At the bottom, a new specialist database link now existing to the mapped entry in AntibioticDB.
The layout of the ligand summary pages has been changed to hopefully provide more of the key information on a ligand at the top of these pages. In the example below we show how this looks for chloroquine.
The main information box now contains:
In addition, the 2D ligand structure is display beside this information in a expandable/retractable section where users can also view physico-chemical properties and SMILES/InChI/InChI Keys.
Previously the top part of the ligand summary pages contained too much white space, repetitions of the ligand name and emphasised the ligand ID, physico-chemical properties and to some extent buried comments, SMILES/InChI keys and useful links (such as to the ligand activity graphs).
The reorganisation of the page now emphasises key information. GtoPdb curator comments in particular contain a valuable description of the ligand and provide explanations as to why they have been curated in GtoPdb.
WHO essential medicines
Any ligands included in the World Health Organization (WHO) Model List of Essential Medicines (21st list, 2019) are now tagged in GtoPdb. This means users can easily view these ligands on our ligand list page:
https://www.guidetopharmacology.org/GRAC/LigandListForward?type=WHO-essential&database=all
It is also possible, as it is for all ligand lists, to download this set from this page by clicking the download link in the top-right of the table (see below).
Both the ligand activity graphs and pharmacology search tools have now been updated to access the latest ChEMBL release (ChEMBL 27).
Links to DrugCentral 2020
We have added links to the DrugCentral 2020 Online Drug Compendium. DrugCentral provides information on active ingredients chemical entities, pharmaceutical products, drug mode of action, indications, pharmacologic action. It monitors FDA, EMA, and PMDA for new drug approval on regular basis to ensure currency of the resource. Limited information on discontinued and drugs approved outside US is also available however regulatory approval information can’t be verified. The database is developed and maintained by Oleg Ursu and Tudor Oprea and the web application is developed by Jayme Holmes.
We have so far mapped 1433 ligands in GtoPdb to entries in DrugCentral.
This post covers three recent publications with a common theme and whose authors are collaborators with GtoPdb, thus making them as a trio particularly suitable for combined review. These are; Discovery of Human Signaling Systems: Pairing Peptides to G Protein-Coupled Receptors [1], Novel approaches leading towards peptide GPCR de-orphanisation [2] , and Advances in therapeutic peptides targeting G protein-coupled receptors [3]. As expected, these are all clearly written, contain valuable detail and each includes its own introduction. Therefore this piece does not need to go over these works per se but simply point out their coverage, domain of application, along with brief explanations of entity linking and/or external look-up options.
For context, it should be pointed out that historical success in de-orphanising GPCRs (i.e. reproducibly pairing them with physiologically-relevant endogenous ligands) has been difficult and slow. This remains so, despite decades of intense effort from not only all major pharmaceutical companies but also academic groups utilising various flavours of de-orphanistaion screening approaches and technologies. This was pursued in parallel with cloning (and patenting) the complete repertoire of human GPCRs as they slowly came into view. Initially done via mining millions of expressed sequence tag (EST) transcripts in the mid 90’s, this later shifted focus to genomic DNA surfacing between appropriately 1998 and 2000. The modest progress even after the completion of the draft human genome can be discerned from the table below compiled in 2000 [4].
Progress ~ 2000- 2018 is outlined both in Concise Guide 2019/20 : G Protein-Coupled Receptors (PMID: 31710717) [5] and in the Guide to Pharmacology latest parings list .
As the first of this trio of papers, Foster et al. [1] have significantly advanced the de-orphanisation field in 2019 by integrating computational and experimental approaches for peptide-GPCR pairings. They began with comparative sequence and structural analyses to gain biological insights into the human peptide-receptor signalling landscape. They were then able to leverage these features to mine candidate peptide ligands from the entire human genome, arranged for these to be synthesised and then tested for activity in a broad range of pairing assays that included known ligands as positive controls. The impressive scope of results is indicated in their Fig 7 (below).
In summary, the 53 peptides with validated receptor-dependent responses first reported in this paper, has expanded the known human peptidergic signaling network from 348 to 407 interactions. It is also important to note that they were unable to reproduce no less than six reported parings in the literature from 2004 to 2016 (i.e. those de-orphanisations could not be verified).
In collaboration with the Copenhagen crew, this paper was curated by the GtoPdb team according to their target mapping stringency and potency thresholds. Consequently, 11 new peptides have been annotated with the quantitative ligand binding data reported for the newly de-orphanised receptors. These are shown below with their PubChem Substance Links (SIDs) for which GtoPdb (so far) has becomes the only source of these compound structures (CIDs) from over 730 submitters.
[画像:Capture]In PubChem these can be accessed by scrolling down to “Related information” on the lower right hand facet of PMID: 31675498 and clicking on “PubChem Substance”. Each entry has a pointer back to GtoPdb from the SID and the nomenclature of the peptides matches that used in the paper. However, users wanting to select these entries from within GtoPdb can go to Ligand Search Tools > Search for data by literature reference > Enter PubMed Id: 31675498 > Select field to search: PubMed ID. This brings back 16 entries since it includes the 11 ligands above plus the five receptors for which binding data was curated (n.b. these instructions apply to the old PubMed interface, so some of this may change for the new interface). While the version of the article in Cell journal itself has no entity outlinks (except for references) the PubMed Central version has automatically linked the PDB IDs. All the GPCR protein/gene names should retrieve in GtoPdb or of course GPCRdb
The second article in the trio is a review by Hauser et al [2]. This covers some of the same ground as above but includes an assessment of contemporary methodologies of de‐orphanisation. The informative Figure 1, summarising extensive data mining, is shown below.
In addition to indicating the ligand status of the GPCRs the circles extend to clinical relevance, disease therapeutic potential, publication density and tool compounds from ChEMBL. Note that while ChEMBL links to compounds with a wide range of potentcies GtoPdb has a more stringent annotation of smaller number of probe candidates including very recent publications (e.g. these 11 ligands above are not in ChEMBL26).
One of the advantages of publishing in BJP is the provision of author-specified out-links to GtoPdb ligands and targets (see PMID 30087946). These have been duly added to the PDF for all GPCRs mentioned in the text as well as any ligands that were already in GtoPdb before this review. Examples for five links are shown below.
Clicking on “GPR15” above thus takes us the full annotation for that receptor, including the agonist ligand records shown below.
Thus GPR15L (71-81) corresponds to SID 404859015. The format of Table 1 unfortunately precluded the addition of the live-links for the new ligands.
The last of this trio is an in-depth review by Davenport et. al. [3] that encompasses translation aspects of peptides that modulate GPCRs into clinically effective drugs. This may even eventually extent to analogues of the peptides from the first two papers perhaps even related to new de-orphanisations. As detailed in this review therapeutic peptides have recently been undergoing a development technology renaissance via half- life extension, stapling and resistance to proteolysis. These measures improve pharmacokinetics and oral bioavailability that have hitherto been much less favourable for peptidic structures in comparison to conventional small-molecule drug candidates. An overview of these peptide approvals and advanced candidates is taken from their Figure 1.
This comprehensive review goes on to describe in detail 26 synthetic peptides approved for clinical use (20 agonists and 6 competitive antagonists) targeting eight class A receptor families. It also includes a useful structural perspectives not only on peptide binding sites but also addresses the frequently overlooked aspects associated with unnatural amino acids and chemical modifications.
It seems somewhat anachronistic that the Nat Rev Drug Discov PDFs are entity link-free zones (although this FAIR-gap has been pointed out to them). Notwithstanding, the GPCR names should retrieve cleanly from GtoPdb and GPCRdb. Most of the peptide names in the review should also retrieve their ligand entries from GtoPdb but these will eventually be cross-checked both for the resolution of equivocalities and addition of new development candidates with referenced binding data.
Constituting a well-referenced overview of the field, these three papers also represent “good news” for peptides and their analogues. Notwithstanding, this slide set outlines some of the challenges for curating peptides and searching them in databases.
**********************************
Comments by Chris Southan, Fellow of the University Edinburgh, Owner of TW2Informatics , Chair of NC-IUPHAR Subcommitees for Proteases and Drug Targets and Chemistry (DRUTACS), ORCID 0000-0001-9580-0446. As declaration of interest (but not conflict!) Chris was pleased to be a co-supervisor for Alex Hauser’s PhD work on Computational Receptor Biology
[1] Discovery of Human Signaling Systems: Pairing Peptides to G Protein-Coupled Receptors. Foster SR, Hauser AS, Vedel L, Strachan RT, Huang XP, Gavin AC, Shah SD, Nayak AP, Haugaard-Kedström LM, Penn RB, Roth BL, Bräuner-Osborne H, Gloriam DE. Cell. 2019 Oct 31;179(4):895-908.e21. doi: 10.1016/j.cell.2019年10月01日0. PMID: 31675498.
[2] Novel approaches leading towards peptide GPCR de-orphanisation. Hauser AS, Gloriam DE, Bräuner-Osborne H, Foster SR. Br J Pharmacol. 2020 Mar;177(5):961-968. doi: 10.1111/bph.14950. Epub 2020 Feb 3. PMID: 31863461.
[3] Advances in therapeutic peptides targeting G protein-coupled receptors. Anthony P. Davenport AP, Scully CCG , de Graaf C, Brown, AJH, Maguire JJ. Nat Rev Drug Discov (2020). https://doi.org/10.1038/s41573-020-0062-z.
[4] The impact of genomics on drug discovery. Beeley LJ, Duckworth DM, Southan C. Prog Med Chem. 2000;37:1-43. PMID: 10845246.
[5] The Concise Guide to PHARMACOLOGY 2019/20 : G Protein-Coupled Receptors. Alexander SPH, Christopoulos A, Davenport AP, Kelly E, Mathie A, Peters JA, Veale EL, Armstrong JF, Faccenda E, Harding SD, Pawson AJ, Sharman JL, Southan C, Davies JA; CGTP Collaborators. Br J Pharmacol. 2019 Dec;176 Suppl 1:S21-S141. doi: 10.1111/bph.14748. PMID: 31710717.
These are some of the ligands under curation (pre-release) in the Guide to Pharmacology. We expect them to be available on the website at the next database release (2020.3).
Ligand ID: 10891
Name: aviptadil
[画像:10891]
Comment: Aviptadil is a vasoactive intestinal peptide analogue, VPAC1 receptor agonist.
The HELM annotation for aviptadil is PEPTIDE1{H.S.D.A.V.F.T.D.N.Y.T.R.L.R.K.Q.M.A.V.K.K.Y.L.N.S.I.L.N}$$$$.
Ligand ID: 10745
Name: MDL-28170
IUPAC: benzyl N-[(2S)-3-methyl-1-oxo-1-[[(2S)-1-oxo-3-phenylpropan-2-yl]amino]butan-2-yl]carbamate
Comment: PRD_002214 is a peptide-like inhibitor of the main protease (MPRO) of SARS-CoV-2. The structure was obtained from the RCSB Protein Data Bank entry 6LU7 which shows the ligand in complex with the protease. The article describing the work has not yet been published (March 12, 2020), but the inhibitor (also referred to as N3) has been deployed in other MPRO crystallisation studies (Yang et al. (2005)).
Ligand ID: 10752
Name: leronlimab
Comment: Leronlimab (PRO140) is a humanized anti-CCR5 (C-C motif chemokine receptor 5) monoclonal antibody [5-6]. It was originally developed by CytoDyn as a novel strategy to manage CCR5-tropic HIV infection.COVID-19: Leronlimab has been administered to a number of patients with severe COVID-19, to determine its potential to reduce cytokine storm caused by this disease.
Ligand ID: 11026
Name: meplazumab
Comment: Meplazumab is a humanized IgG2 monoclonal antibody that targets CD147 (basigin). It is being developed by Jiangsu Pacific Meinuoke Bio Pharmaceutical.
COVID-19
SARS-CoV-2 may utilise CD147 as an additional entry receptor to infect host cells [2]. Meplazumab can block this interaction between CD147 and SARS-CoV-2 spike protein.
We are pleased to have been able to make an expeditious database release (version 2020.2), following on from our last update in March 2020 in order to make public new curation specifically related to SARS-CoV-2.
The Guide to Pharmacology coronavirus information page continues to be updated on a regular basis to capture the latest pharmacological strategies under investigation to mitigate against COVID-19.
These are some of the ligands under curation (pre-release) in the Guide to Pharmacology. We expect them to be available on the website at the next database release (2020.2 – no earlier than April 2020).
We are pleased to announce our first database release of 2020 – version 2020.1.
We have added a new coronavirus page to the Guide to Pharmacology which aims to consolidate key pharmacological information and resources related to coronavirus. We have gathered information from various sources, with an aim to cover as many of the pharmacological strategies being considered as we could find. Links are provided to the key ligands and target pages in GtoPdb (where data is available). The page also provides other useful links and resource. We will aim to keep this page updated in this fast changing situation.
GtoPdb contains a couple of useful tools for searching and visualising pharamcology data – that combine data from ChEMBL with GtoPdb data. In this release we’ve update our site to make accessing the visualisation charts easier
Ligand Activity Charts
This tool provides charts with box plots summarising all the activity data for a ligand taken from ChEMBL and GtoPdb across multiple targets and species. Separate charts are created for each target, and where possible the algorithm tries to merge ChEMBL and GtoPdb targets by matching them on name and UniProt accession, for each available species. However, please note that inconsistency in naming of targets may lead to data for the same target being reported across multiple charts.
Previously, access to the charts was only found on a ligand’s summary page, under the biological activity tab. We’ve now moved that link so it appears more prominently on these pages.[画像:ligSumLink]
We have also added a new icon on the ligand list pages which indicated whether there are activity charts available for the ligand. Click the icon takes the users straight to the charts.
New ‘bar-chart’ icon added to ligand list to indicate availability of activity charts
We have published a review of the Guide to Immunopharmacology in Immunology at the start of the year. The review provides a deeper context for how the resource can support research into the development of drugs targeted at modulating immune, inflammatory or infectious components of disease.
Harding, S. D., Faccenda, E., Southan, C., Pawson, A. J., Maffia, P., Alexander, S., Davenport, A. P., Fabbro, D., Levi-Schaffer, F., Spedding, M., & Davies, J. A. (2020). The IUPHAR Guide to Immunopharmacology: connecting immunology and pharmacology. Immunology, 10.1111/imm.13175. Advance online publication. https://doi.org/10.1111/imm.13175
GPCRs:
Ion Channels:
NHRs:
Enzymes and Other Protein Targets:
Guide to Malaria Pharmacology Updates:
The ligand summary pages have been updated to improve the display of clinical trials information. Key clinical trials involving the ligand are now displayed in a separate table, under the clinical data tab. The table includes link out the clinical trial plus trial title, type, source, curator comments and references.
Approved drug with primary targets download
A new download file of all approved drugs and their primary targets has been added to the download page.
Melatonin targets two high-affinity receptors, MT1 and MT2, that belong to the G protein-coupled receptor (GPCR) superfamily (1,2). Drugs acting on melatonin receptors are subscribed for circadian disorders (jet lag, shift work, etc.), insomnia and major depression (3). All marketed drugs are non-selective agonists targeting both receptor types so far. Despite large chemical synthesis campaigns over the last 20 years, the pharmacology of melatonin receptors remains poorly explored with few inverse agonistic and signaling pathway-biased compounds and few selective compounds, in particular for the MT1 type (2,4). This lack of pharmacological tools goes along with a rather poor diversity in terms of chemical scaffolds currently available (5). Put into this context, a virtual screening looked like a very promising approach to increase the diversity of chemical scaffolds for melatonin receptors, especially now that the crystal structure of the MT1 receptor has been solved (6). The outcome of the virtual screening study performed by Stein et al. (7) met these high expectations. Overall, 15 new chemical scaffolds were identified from the primary docking of over 150 million virtual molecules (7). Some of the primary hits had pEC50 values around 1nM, which is remarkable. In terms of functional properties, primary hits and derivatives showed agonistic and inverse agonistic properties with or without selectivity for MT1 and MT2. Two MT1 selective inverse agonists were further validated in vivo regarding their effects on circadian rhythm parameters in mice under free-running conditions, as well as using an experimental re-entrainment protocol corresponding to an ‘east-bound’ jet lag paradigm.
This study is not only of great value for the melatonin receptor field but will also encourage virtual screening approaches on other GPCRs for which high-resolution structures are available. Compared to other GPCR structures, the MT1 template structure has several limitations as it was in the inactive form despite the presence of the 2‐phenylmelatonin agonist, and it was heavily modified with 9 point mutations and several truncations to facilitate expression and stability (7,8). However, a favorable feature of MT1 for virtual docking might be its ligand binding pocket which is rather small and hydrophobic with only two key residues important for high-affinity binding. This limits the number of poses to be screened by virtual docking. Moreover, although the screening was designed to identify MT1 selective compounds, positive hits included also MT2 selective and non-selective compounds suggesting that virtual screening is currently unable to predict type selective compounds. This might also depend on the quality of the receptor structure and the degree of divergence between receptor types.
In conclusion, this article is a nice example of successful virtual screening on GPCRs, one of the desired benefits of crystal structures, that will accelerate the drug development process towards compounds with improved efficacy and target-selectivity.
This study investigated two MT1-selective inverse agonists, UCSF7447 and UCSF3384, and a selective MT2 agonist, UCSF4226, for their affinities.
Comments by Ralf Jockers, Institut Cochin, CNRS, INSERM, Université de Paris, Paris, France, Chair for NC-IUPHAR Subcommitee for Melatonin receptors.
Thirty eight years after the discovery that injection of the catalytic subunits of cyclic AMP-dependent protein kinase A (PKA) into isolated pig ventricular cardiac myocytes (1) increases the amplitude of L-type Ca2+ current (today known to be formed by Cav1.2 channels, (2)), we are now getting close to understand the responsible molecular mechanism. Initial research that has focused on direct regulatory mechanisms, such as the activation by the α-subunit of the stimulatory heterotrimeric G-protein Gs (3), and direct phosphorylation of the pore-forming α1- or accessory β2-subunit of the Cav1.2 channel has not provided a conclusive answer. β-adrenergic modulation was not (α1 Ser-1928, (4)) or only partially (α1 Ser-1700/Thr-1704, (5)) prevented in mutant mice lacking predicted phosphorylation sites in α1 or β2 subunits (6). This was further supported by an elegant transgenic approach showing that expression of channel complexes with all PKA consensus sites mutated to alanine in the Cav1.2 α1 and the β2 subunits (7) has no effects on β-adrenergic modulation.
Assuming that other proteins may be involved in this regulatory pathway, Liu et al. (7) used a biotinylation-based proteomic proximity assay to identify proteins in the Cav1.2 neighborhood regulated by isoproterenol treatment in cardiomyocytes. This led to the discovery of the small Ras-like G protein Rad as a potential candidate. It belongs to the family of the Rad, Rem, Rem2 and Gem/Kir (RGK) Ras-like GTP-binding proteins, which were shown earlier to inhibit high-voltage-activated Ca2+ channels by binding to their β-subunits. Indeed, Rad appears to be the long-sought missing piece in the puzzle: in HEK293T Rad-mediated inhibition of cardiac Cav1.2 channels was relieved by PKA phosphorylation. Modulation was prevented either by the simultaneous mutation of the known cardiac PKA phosphorylation sites of Rad to alanine or by inhibiting the interaction of Rad with the β-subunit.
This mechanism may be universal and may also explain the PKA modulation of other types of β-subunit associated high-voltage activated Ca2+ channels in Rad expressing cells. It also provides evidence that the mechanism of PKA modulation of Cav1.2 Ca2+ channels appears to differ in different cell types. Clearly, direct phosphorylation of Ser-1928 of the α1-subunit is not required for β-adrenergic regulation of Cav1.2 in the heart (4,7). However, this residue is required for β-adrenergic stimulation of Ca2+ channels in hippocampal neurons and for hyperglycaemia-induced stimulation of Ca2+ currents in arterial smooth muscle cells (8,9).
Despite compelling evidence for this novel mechanism, several questions remain: First, what is the role of Rad-dependent modulation of the cardiac L-type channel in intact cardiomyocytes, for example in cardiomyocytes expressing Rad with mutated PKA phosphorylation sites. Second, what is the relative importance of Rad versus modulation by direct phosphorylation of Ser-1700 (and Thr-1704), which has also been found to confer some of the PKA activation of L-type current in cardiomyocytes of Ser1700Ala mutant mice (5).
Despite these open questions, we are now getting close to understand the molecular mechanisms underlying the fight-or-flight response.
Comments by Jörg Striessnig, University of Innsbruck, Chair for NC-IUPHAR Subcommitee for Voltage-gated calcium channels, Liaison for NC-IUPHAR subcommittees on Voltage-gated ion channels
Two new reports cover further aspects of cannabinoid receptor binding as defined using cryo-EM. These expand on previous reports of the structure of the human CB1 cannabinoid receptor with the antagonists taranabant and AM6538 bound, and with the agonists MDMB-FUBINACA, AM11542 and AM841 bound. Previously reported also is the structure of the CB2 cannabinoid receptor with the antagonist AM10257 bound.
In the first report (1), a non-selective, high potency agonist WIN55212-2 was bound to a complex of the CB2 cannabinoid receptor with Gi to "complete the set" of agonist- and antagonist-bound versions of CB1 and CB2 receptors. The authors suggest marked similarities in the binding configuration of the agonist WIN55212-2 and the antagonist AM10257. Based on these structures, two novel ligands were generated; one predicted to be an agonist and the other an antagonist. The functional data supported these predictions, although both novel compounds were markedly less potent than their respective positive controls.
In the second report (2), agonist-bound versions of both CB1 and CB2 cannabinoid receptors complexed with Gi were also described to ‘complete and expand the set’. AM12033 is a novel nitrilodimethylheptyl analogue of THC with high affinity at both receptors. Structures using this compound were compared to CB1 receptors bound to the THC-like AM841. The authors suggest similar modes of interaction for agonists at the two receptors, supported by numerous examples of non-selective high potency agonists.
Modelling of the second intracellular loop in both reports suggested a structural basis for the CB2 receptor to couple poorly to Gs. Indeed, a single amino acid variation (L222 in CB1 and P139 in CB2 receptors) was highlighted in both studies and evidenced in the previous literature (https://www.ncbi.nlm.nih.gov/pubmed/23667597).
Comments by Prof. Steve Alexander (@mqzspa), University of Nottingham, UK
(1) Xing C. et al. Cryo-EM Structure of the Human Cannabinoid Receptor CB2-G i Signaling Complex. Cell (2020) doi:10.1016/j.cell.202001007 [PMID:32004460]
(2) Hua T. et al. Activation and Signaling Mechanism Revealed by Cannabinoid Receptor-Gi Complex Structures. Cell (2020) doi:10.1016/j.cell.202001008 [PMID:32004463]
The new respiratory coronavirus from Wuhan in China is causing intense research activity in the world. Viruses are evidently associated with respiratory infections ranging in severity from the common cold to pneumonia and death. More than 200 viral types have been associated with the common cold, of which 50% of infections are rhinovirus, but also respiratory syncitial virus, influenza – and coronaviruses, particularly human coronavirus-229E (HCoV-229E), which is ‘relatively benign’. Monocytes are relatively resistant to HcoV-229E infection, but rapidly invades, replicates in, and kills dendritic cells within a few hours of infection (Mesel-Lemoine et al., 2012) [1]. Dendritic cells are the sentinel cells in the respiratory tract, and plasmacytoid dendritic cells are a crucial antiviral defence via interferon production. Thus, these viruses can impair control of viral dissemination and the formation of long-lasting immune memory. The cellular receptor for HcoV-229E is CD-13 (aminopeptidase-N).
The cellular receptors for coronaviruses are critical for cell entry. Severe acute respiratory syndrome (SARS) coronavirus (SARS-CoV), and HCoV-NL63, bind to angiotensin converting enzyme2, ACE2, on ciliated cells in the respiratory tract, the crystal structure is known (Song et al., 2018) [2]. The SARS-CoV spike (S) glycoprotein (with S1 and S2 sub-units) on the outer envelope binds to ACE2, fusing viral and cellular membranes, triggering conformational transformations. Cleavage of the S1/S2 subunits by proteases is critical. Indeed, Simmons et al (2005) [3] showed that SARS-CoV infection involved receptor binding, conformational changes receptor binding and subsequent cathepsin L (and type II transmembrane serine proteases) proteolysis within endosomes – with inhibitors of cathepsin L preventing viral entry. SARS-CoV may lead to markedly elevated cytokine levels, leading to tissue damage, pneumonitis, and acute respiratory distress syndrome (ARDS). The spike proteins in SARS-CoV and Middle East respiratory syndrome coronavirus (MERS-CoV) are crucial for host specificity and jumping between species, e.g. from bats to humans (Lu et al., 2015) [4], and also the recent cross-over to swine acute diarrhoea syndrome coronavirus (SADS-CoV) to pigs (Zhou et al., 2018) [5]. There is intense interest in the complicated ways whereby coronaviruses engage with their target receptors, are activated and replicate. The rapid evolution of coronaviruses is critical as humans have little or no pre-existing immunity SARS- or MERS-CoV.
ACE2 has been identified as a potential receptor for the novel coronavirus that was identified as the cause of the respiratory disease outbreak in Wuhan in late 2019 (referred to as 2019 – nCoV by the WHO, or Wuhan coronavirus) [6,7 & 9]. 2019-nCoV is a betacoronavirus, and, in common with the SARS-CoV [8], the viral spike protein is postulated to engage ACE2 for viral entry [7]. The data presented by Letko and Munzter [7] is preliminary and has not been peer-reviewed, but a rapid pace of research is needed in the present circumstances. For example, in another recent, preliminary report, ACE2 receptor expression is highly expressed in type II alveolar cells (AT2) with much individual variation and the authors reported that the cells also expressed many genes involved in viral reproduction and transmission [10]. While these findings are preliminary, the rapid publications in bioRxiv and similar archives are crucial in what is now officially a global health emergency. This report is a hot topic in the IUPHAR/BPS Guide to PHARMACOLOGY (https://www.guidetopharmacology.org), and further underlines the importance of immunopharmacology and the IUPHAR Guide to IMMUNOPHARMACOLOGY (https://www.guidetoimmunopharmacology.org/immuno/index.jsp).
Comments by Prof. Michael Spedding, Spedding Reseach Solutions; Prof. Steve Alexander (@mqzspa), University of Nottingham; Dr. Elena Faccenda, University of Edinburgh; and Prof. Francesca Levi-Schaffer, The Hebrew University of Jerusalem.
Note from bioRxiv: bioRxiv is receiving many new papers on coronavirus 2019-nCoV. A reminder: these are preliminary reports that have not been peer-reviewed. They should not be regarded as conclusive, guide clinical practice/health-related behavior, or be reported in news media as established information.
Nieng Yan’s group has now published the cryo-EM structure of the selective T-type Ca2+ channel blocker Z944 bound to the pore-forming α1-subunit of Cav3.1 T-type Ca2+ channels (1). This nicely adds to recent publications reporting of the high-resolution structures of L-type Ca2+ channel blockers (nifedipine, nimodipine, amlodipine, verapamil, diltiazem) bound to the rabbit Cav1.1 skeletal muscle Ca2+ channel (2) and to the model Ca2+ channel CavAb (Ca2+-selectivity engineered into the bacterial homotetrameric Na+-channel NavAb; 3, 4).
Together these three publications provide important insight into how small molecule channel blockers interact with their high affinity binding domains within Ca2+ channels. They also reveal a remarkable similarity between the core structures (i.e. the voltage-sensors and the pore domains) between not only the closely related Cav1.1 and Cav3.1 channels but also their bacterial ancestor CavAb.
Moreover, although some differences exist, the binding poses of L-type Ca2+ channel blockers in the mammalian Cav1.1 and the bacterial CavAb channel are very similar. In both structures verapamil and diltiazem bind within the central cavity, compatible with their pore-blocking properties predicted in functional studies. In contrast, dihydropyridines like nimodipine and nifedipine bind within a fenestration created by adjacent transmembrane S6 helices (IIIS6 and IVS6). These fenestrations penetrate the sides of the central cavities in both voltage gated Na+– and Ca2+ channels and connect them to the surrounding lipid bilayer of the plasma membrane (5). In accordance with functional studies, these dihydropyridines must therefore inhibit L-type channels by allosterically interfering with gating rather than pore block.
These findings raise the important question about how Z944 can selectively inhibit T-type Ca2+ channels. This drug is currently in clinical development for the treatment of epilepsy and neuropathic pain. It appears that Z944 bound to Cav3.1 combines the binding modes of both pore blockers and allosteric blockers. Its phenyl ring projects into the fenestration between repeats II and III (similar to nifedipine binding), whereas the other end of the molecule projects down through the central cavity to the intracellular mouth of the pore. A prominent interaction was found with lysine-1462, a residue unique for T-type channels. Its mutation to L-type residues reduced drug sensitivity in functional studies suggesting that it is one of the determinants for T-type selectivity.
Taken together the accumulating structural insight into drug binding and modulation will greatly facilitate structure-guided drug discovery. This may lead to new oral subtype-selective blockers of voltage-gated Ca2+ channels with therapeutic potential for the treatment of epilepsy (T-type), pain (T-type, N-type), tremor (T-type), primary aldosteronism (Cav1.3 L-type), progression of Parkinsons disease (Cav1.3, R-type) or rare neurodevelopmental disorders (Cav1.3 L-type) (6,7).
Comments by Jörg Striessnig, University of Innsbruck, Chair for NC-IUPHAR Subcommitee for Voltage-gated calcium channels, Liaison for NC-IUPHAR subcommittees on Voltage-gated ion channels
Leukotrienes are lipid mediators of inflammation, initially recognized for their role in asthma, but also having potent effects in for example cardiovascular and neurological diseases as well as in cancer. The initial pharmacological classification of leukotriene receptors based on antagonist selectivity in smooth muscle was shown to be valid at the gene and protein levels. Following the recent description of the CysLT1 receptor structure, Gusach et al. now describe the crystal structures of the CysLT2 receptor in complex with dual CysLT1/CysLT2 receptor antagonists (1). This is an important advancement, since it allows to reveal specific characteristics of the two CysLT receptors in terms of ligand recognition and subtype selectivity. In this context, differences in the intracellular helix 8 emerge as particular subtype determinants, which may affect G-protein regulation and β-arrestin binding. The study by Gusach et al. (1) further provides structural insights into the changes in ligand binding and downstream signaling by CysLT2R disease-related single nucleotide polymorphisms (SNP) associated with asthma and cancer. The deciphering of the crystal structures for the CysLT1 and CysLT2 receptors will open up for the design of CysLT receptor antagonists with improved affinity/efficacy or subtype selectivity profiles. The insights into the CysLT receptor genome-structure-function relations may facilitate the prediction of disease associations and potentially further expanding the therapeutic indications of CysLT receptor antagonists.
Comments by Magnus Bäck, MD PhD, (@TransCardio ), Karolinska Institutet and University Hospital, Stockholm, Sweden; Chairman NC-IUPHAR subcommittee on Leukotriene Receptors
(1) Gusach, A., Luginina, A., Marin, E. et al. Structural basis of ligand selectivity and disease mutations in cysteinyl leukotriene receptors. Nat Commun 10, 5573 (2019) doi:10.1038/s41467-019-13348-2 [PMID:31811124]
Muscarinic receptors consist of 5 G protein-coupled receptors which along with nicotinic ion channels mediate the effects of acetylcholine. Despite years of research on the role of muscarinic receptors in the brain and periphery the Muscarinic M5 receptor has stood out in contrast to M1 -M4 as one which we know little about. The M5 receptor has a unique and unusual distribution in the brain being enriched in mid brain dopamine neurons and in the cerebellum. A lack of selective chemical tools has hampered our ability to study the function of the receptor. Now Vuckovic et al. [1] have solved the X-ray crystal structure of the M5 receptor in complex with the non-selective antagonist tiotropium in the presence of a conformational stabilizing mutation. Since tiotropium was also used to solve the structure of several other muscarinic receptors a direct comparison can be made across the subtypes which will enable the design of selective tool compounds. Significant progress has been made in utilizing crystal structures to design selective orthosteric and allosteric ligands at this family of receptors. This final structure completes the set of muscarinic receptor structures solved and hopefully will pave the way to further understanding of the biology of M5 and its role in disorders such as drug addiction and depression.
Comments by Fiona H. Marshall, Discovery Research UK, MSD (@aston_fm)
(1) Vuckovic Z et al. (2019). Crystal Structure of the M5 Muscarinic Acetylcholine Receptor. Proc Natl Acad Sci USA, DOI: 10.1073/pnas.1914446116. [PMID: 31772027]
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
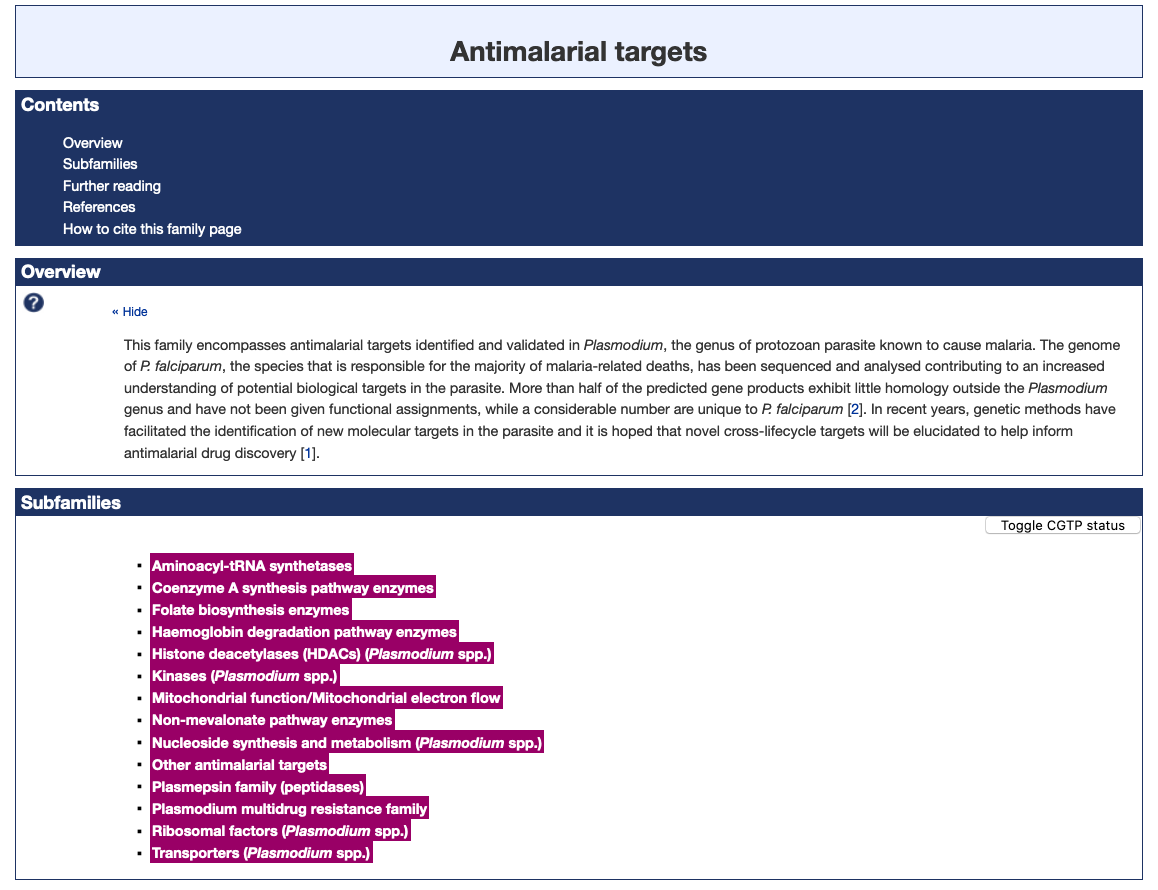{kind=link}
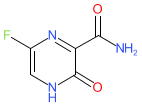{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}